1.Introduction
Cellular immunity is orchestrated by T cells through their immense T-cell receptors (TCRs) repertoire, which interact with antigenic peptides presented by major histocompatibility complex (pMHC) molecules. Here, we present DeepTR, a one-stop collection of unsupervised and supervised deep learning approaches for pan peptide-MHC class I binding prediction, antigen-specific TCR clustering, and T cell response specificity prediction achieved by mimicking crucial steps of the antigen presentation pathway. DeepTR implemented comprehensive featurization strategies to numerically embed sequences of antigens, MHC molecules and six TCR loops of TCR repertoires in a biologically meaningful manner. We demonstrated that DeepTR yields higher predictive performance and more efficient feature representation for peptide binding to MHC (pMHC module) and enables superior antigen-specific TCR featurization upon TCR module than current state-of-the-art approaches. In addition to accurately predicting T cell activation by leveraging the trained numeric embedding from pMHC and TCR modules through a transfer learning strategy, DeepTR also enables the discovery of specific TCR groups upon different binding modalities and further identifies the conserved gene usage and complementarity determining region 3 (CDR3) motifs, including novel subgroups of TCRs via a new mechanism. Finally, structural analyses strongly supported that DeepTR can characterize important contact residues that mediate TCR-antigen binding specificity and make functionally relevant and biologically meaningful predictions. DeepTR may advance our understanding of the mechanisms of T cell-mediated immunity and yield new insight in both personalized immune treatment and development of targeted vaccines.
2. Model
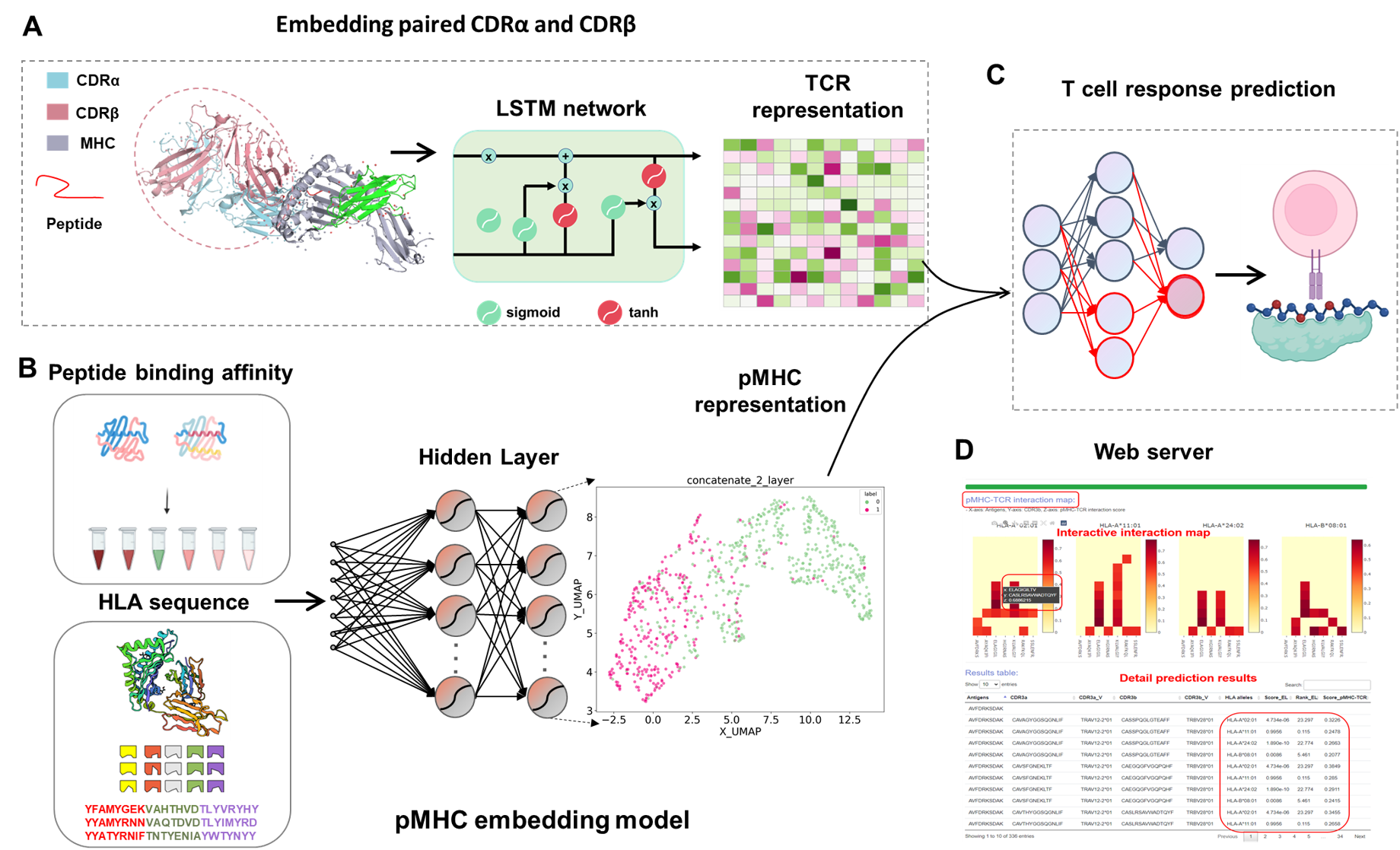
Figure 1. The workflow of DeepTR for T cell response prediction.
First, for embedding of eptide antigen presented by MHC (pMHC), we developded a new pan-allele MHC class I predictor through the deep neural network based on over 200,000 quantitative binding affinity measurements. The input of this model is the MHC class I sequences and the antigen peptides. The layers before output should contain important information regarding the overall structure of the pMHC complex, which could provide high-quality MHC-antigen co-embedding. Moreover, to improved TCR repertoires featurization, anohter deep LSTM autoencoder model for learning TCR embeddings was implemented. We encode >20,000 paired α/β TCR sequence data and their six complementary determining regions (CDRs) into high-dimensional physicalchemical feature representations. Finally, we leveraged the trained numeric vector encodings of TCRs and pMHCs for learning the pairing between them. We constructed a fully connected deep-learning network based on the output of these two submodels, leading to a final layer with a single neuron for predicting the pairing.
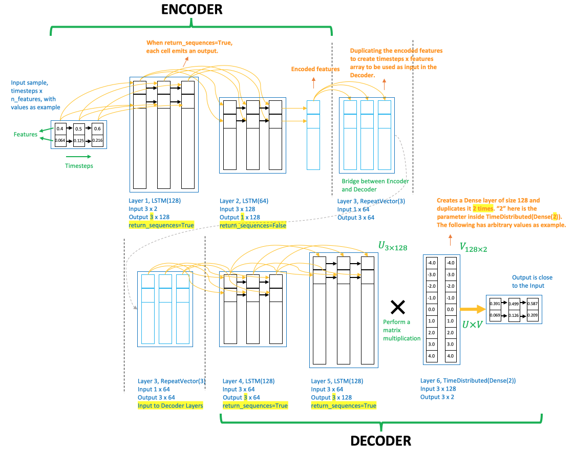
Figure 2. The architecture of long short-term memory (LSTM) autoencoder.
3. Model performance
3.1 pMHC module
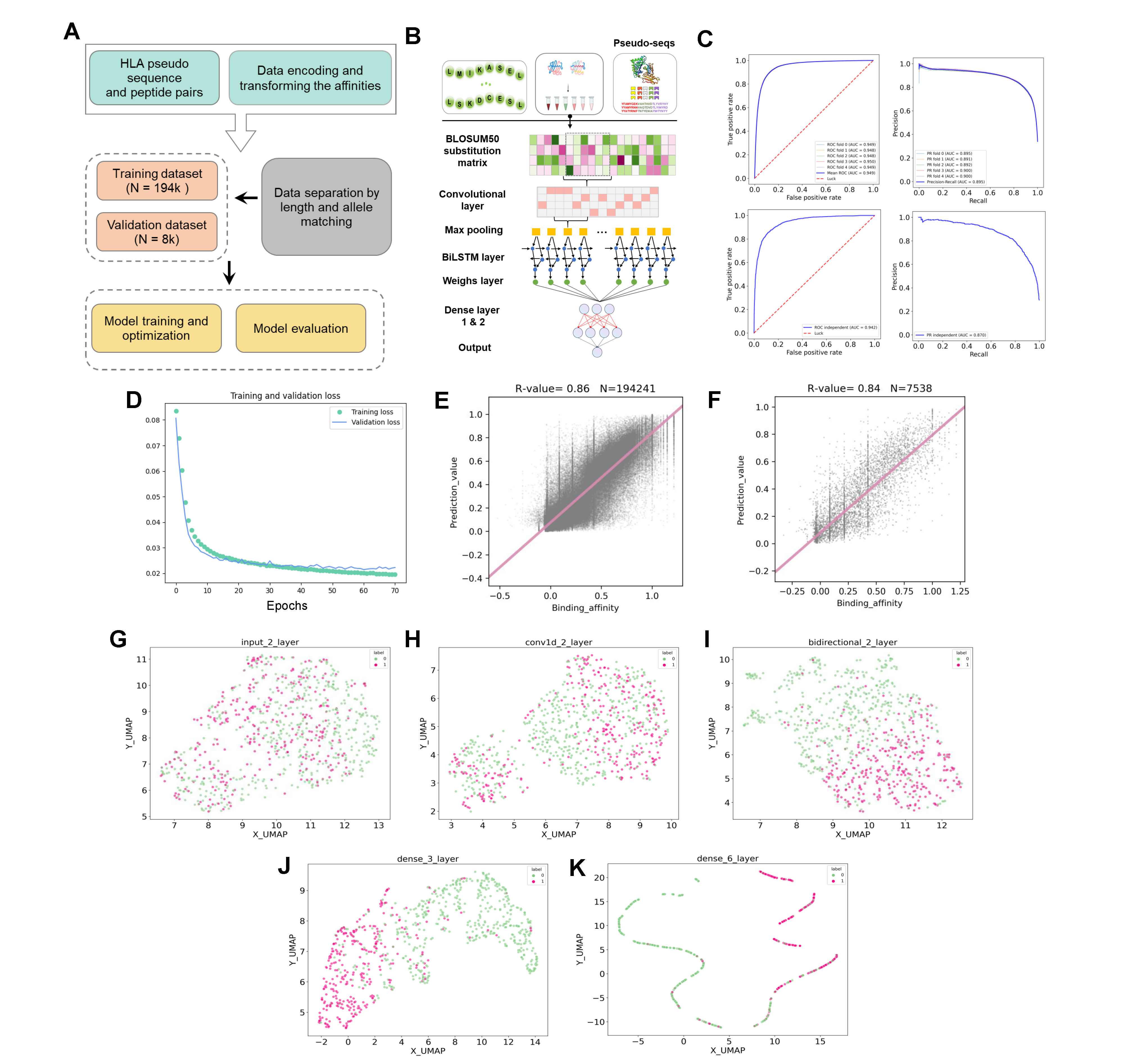
Figure 3. Implementation and validation of pMHC module.
To access the prediction performance of Net-pMHC, a five-fold cross-validation was performed on the curated binding affinity dataset. The receiver operating characteristic (ROC) curves were drawn and the corresponding AUC values were calculated. As a result, when applying this hybrid deep learning architecture, the Net-pMHC, which automatically learns discriminative features and essential residues from the peptides along the layer hierarchy, performed well with the average AUC values of 0.950 from five-fold CV. In addition, Net-pMHC also achieved a high AUC value of 0.942 when using the independent test dataset, indicating the adaptability of our model. We also calculated PCC between predicted and quantitative binding affinities to further evaluate the predictive ability of our model. Net-pMHC achieved PCCs of 0.88 and 0.84 in five-fold CV and independent testing, respectively, a good improvement when compared to the previously reported netMHCpan (BA option) with PCC as 0.76. As above, Net-pMHC was accurate and robust for the peptide-MHC binding prediction, in light of consistent and promising AUC and PCCs in both five-fold CV and independent testing. Although the output layer is dedicated to predicting antigen binding to MHC, the layers before it contain important information regarding the overall structure of the pMHC complex. Thus, we visualized the pMHCs for several well characterized MHC alleles, e.g., HLA-A*02:01, using UMAP method based on the feature representation generated from different network layers. We found our model could hierarchically learn a more efficient and interpretive feature representation of the pMHCs. More specifically, the predicted features for pMHCs and non-pMHCs were mixed at the input layer. However, as the predicted features passed through the CNN and LSTM layers, the model began differentiating between pMHCs and non-pMHCs. The attention layer could distribute higher weights to the essential positions for pMHCs prediction. When integrating with the output from the LSTM layer, pMHCs and non-pMHCs tended to separated clearly, indicating this method could efficiently infer feature representation. Overall, we demonstrated that the Net-pMHC could provide accurate pan peptide-MHC class I binding prediction and generate representative immediate numeric embedding of binding preference for pMHCs.
3.2 TCR module
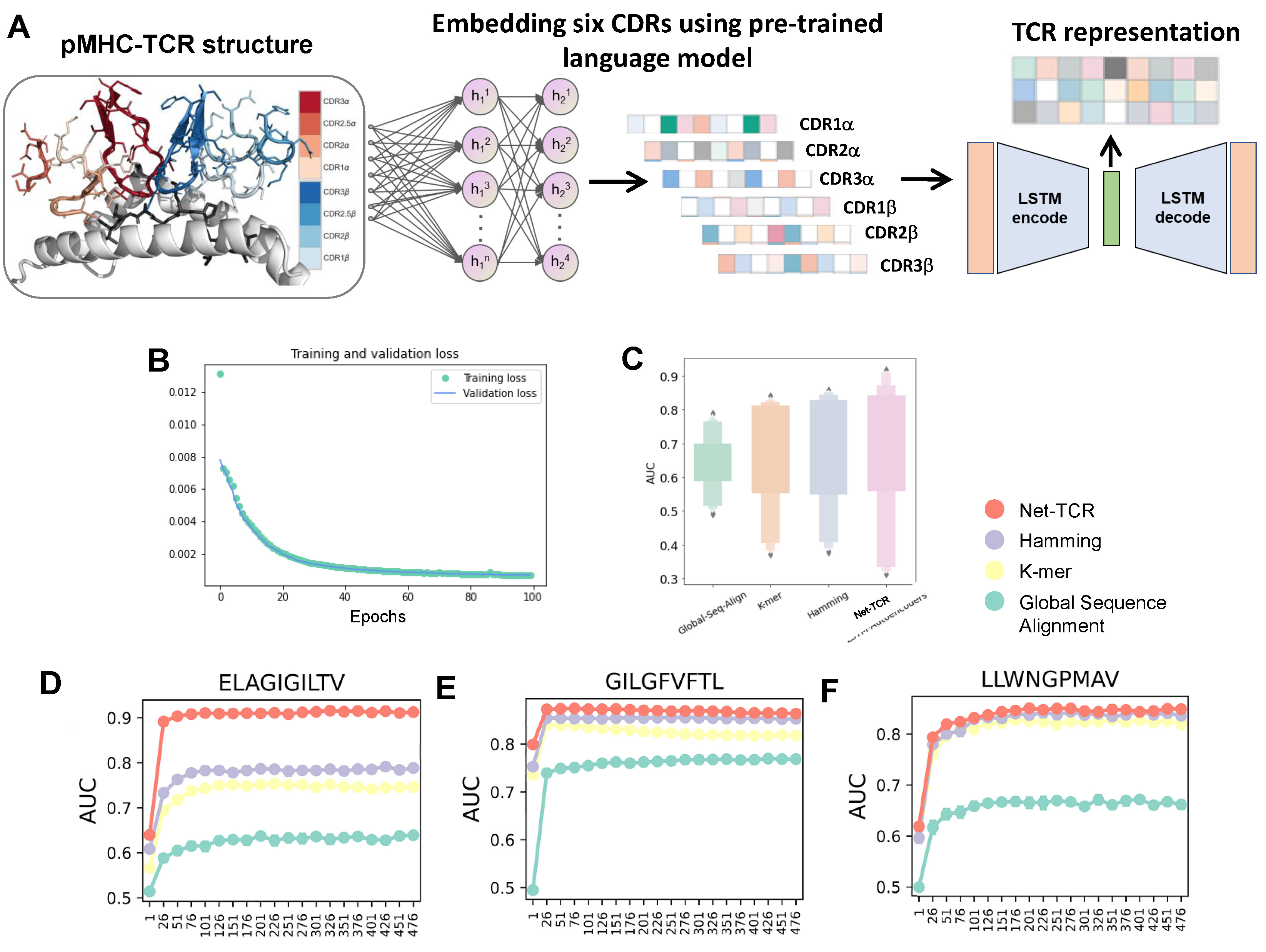
Figure 4. Development and comparison of TCR modules.
The unsupervised learning strategy was implemented to capture the underlying features from a high-dimensional space with the motivation of clustering TCR sequences likely binding to the same antigen. To further evaluate the capability of Net-TCR for TCR featurization, we compared it with some other commonly used methods, such as hamming distances, K-mer representation and global sequence alignment. For example, both GLIPH and TCRdist utilized Hamming distances as the core clustering engine, and ImmunoMap applied the strategy of global sequence alignment . We selected all antigen-specific pMHCs which contain at least 100 distinct α/β TCR sequences from our curated dataset. These pMHCs could serve as ground truth for evaluating antigen specificity. TCR distances from the various featurization methods (Net-TCR, K-mer, hamming distances and global sequence alignment) were calculated. A K Nearest Neighbors (KNN) algorithm across a wide range of K (from 1 to 500) was implemented to determine whether the above TCR featurization methods can accurately distribute a TCR sequence to corresponding antigen using a five-fold CV strategy. AUC values for each method were calculated. The results showed that Net-TCR achieved higher performance and outperformed the current state-of-the-art approaches. More specifically, we further evaluated our model for correctly clustering TCR sequences of the same specificity on some well-studied pMHC, such as ELAGIGILTV-MART1, GILGFVFTL-Flu-MP, LLWNGPMAV-NS4B. Net-TCR classified the TCRs of ELAGIGILTV-MART1 with an average AUC of 0.91, leading to an over 22% AUC improvement compared to other methods. Net-TCR also showed better clustering ability of antigen specificity on the other two pMHCs. These results demonstrated that Net-TCR, which incorporates transfer learning strategy and LSTM autoencoder, was able to comprehensively feature antigen-specific TCR sequences.
3.3 pMHC-TCR module
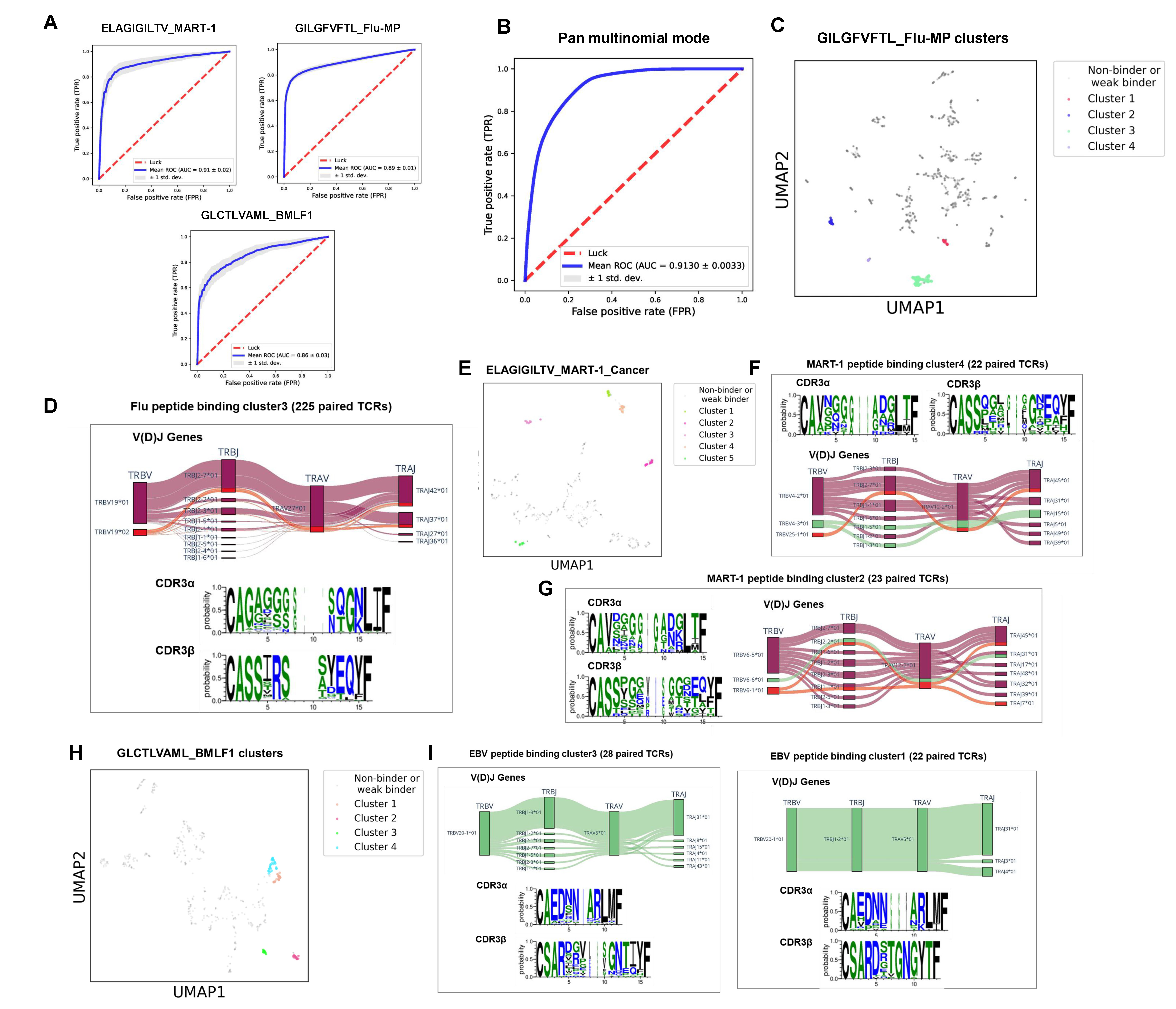
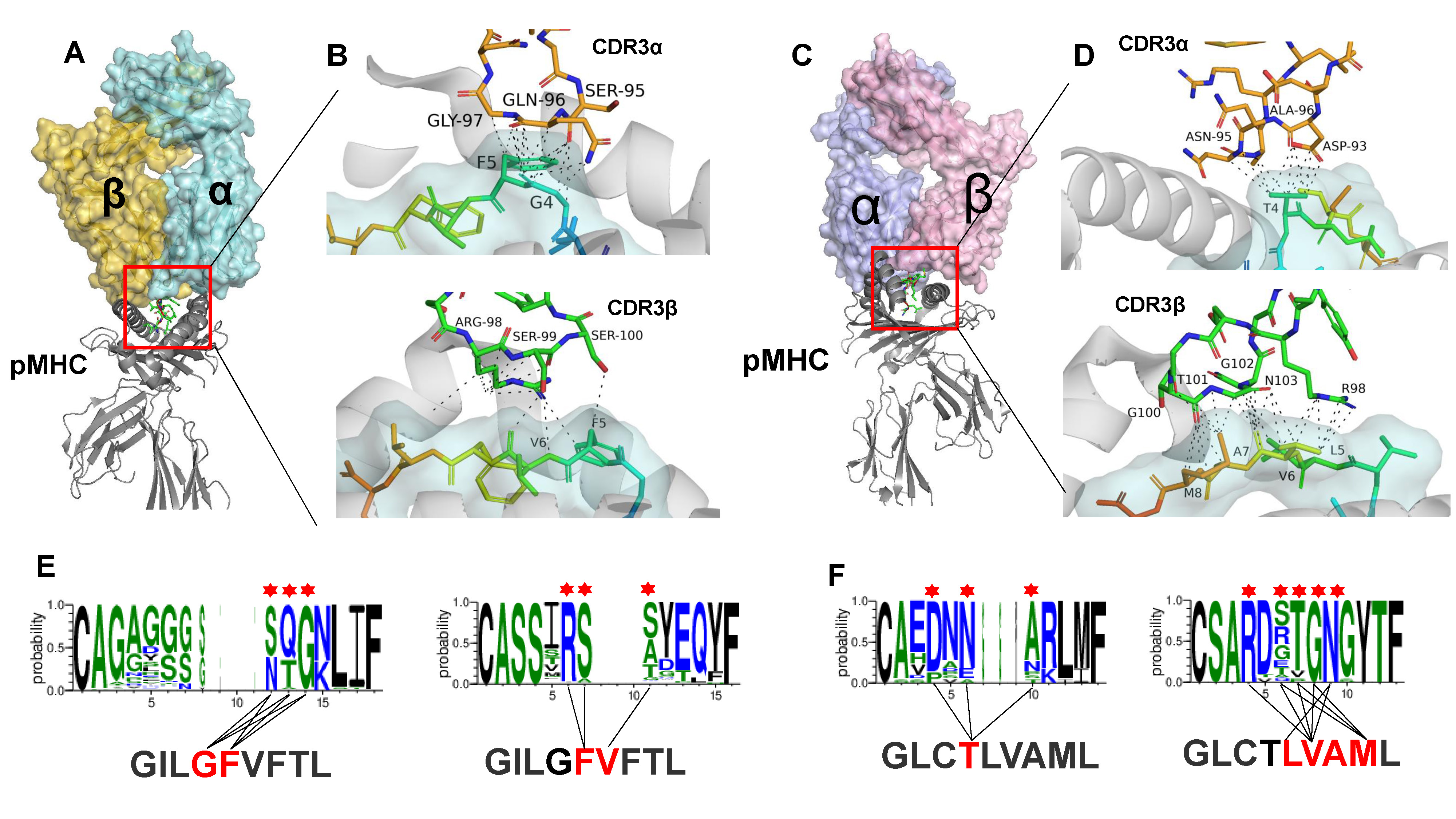
Figure 5. Performance and characterization of pMHC binding TCRs by DeepTR.
A bootstrapping strategy was implemented for training TRpred model and evaluating its performance. Ten independent five-fold stratified CV were performed. Each of the ten iterations considered random splits of the training sets. The mean and standard deviation of the AUC values across all iterations represent the performance of the classifier. For some well-studied pMHCs, e.g., ELAGIGILTV-MART1, GILGFVFTL-Flu-MP, GLCTLVAML-BMLF1, TRPred (binomial mode) could distribute the TCRs to these pMHCs with an average AUC of 0.91, 0.89, and 0.86, respectively. The results demonstrated that the joint supervised model can effectively mimic the process of antigen presentation for T cell activation prediction. However, the known antigenic peptides that TCRs specifically bind to are currently very limited. This makes the existing binomial methods incapable of predictig TCR binding specificities of neoantigens, which is central to understanding cancer progression, prognosis, and responsiveness to immunotherapy. We thus extended the TRPred model with multinomial option for pan TCR-neoantigen binding prediction. As a result, TRPred (multinomial mode) achieved high performance with the average AUC values of 0.91 with five-fold CV, indicating promise for the characterization of TCR binding profiles of neoantigens.
4. General webserver pipeline:
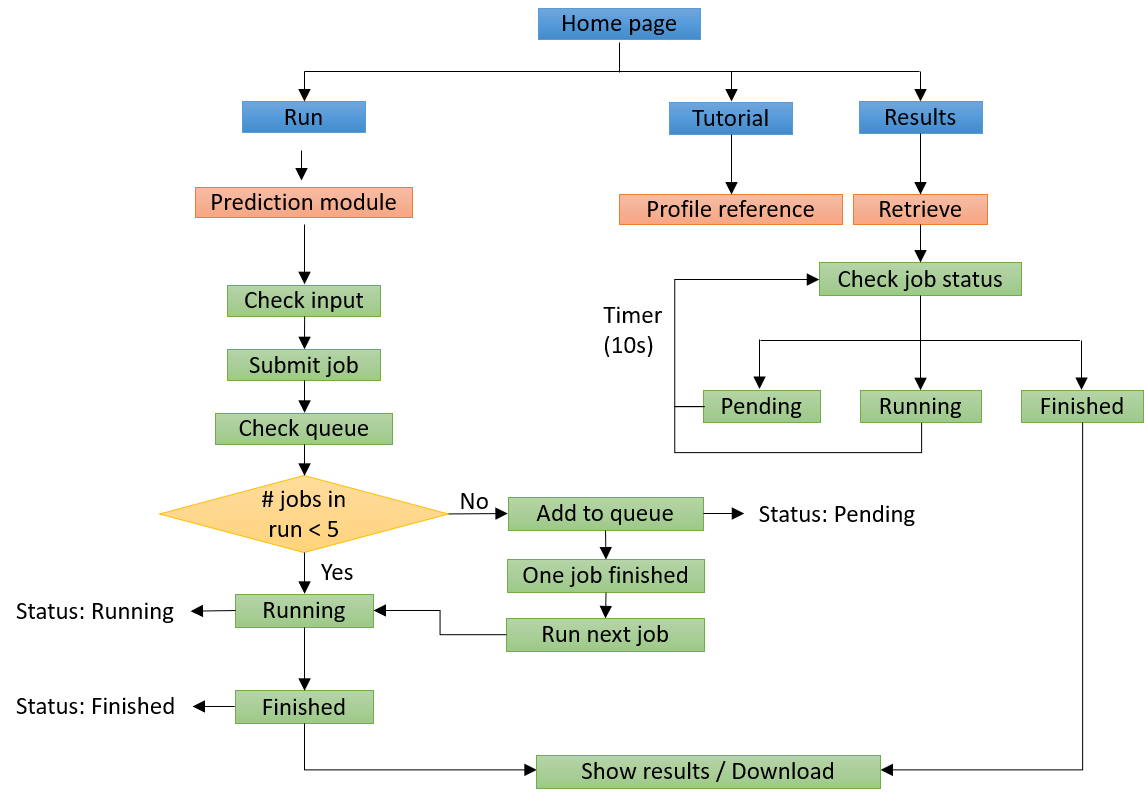
Figure 4. General webserver pipeline for DeepTR server.
Our webserver provides user-friendly interfaces for users to submit jobs, check job status, and retrieve results.
5. Probing T cell response:
Input
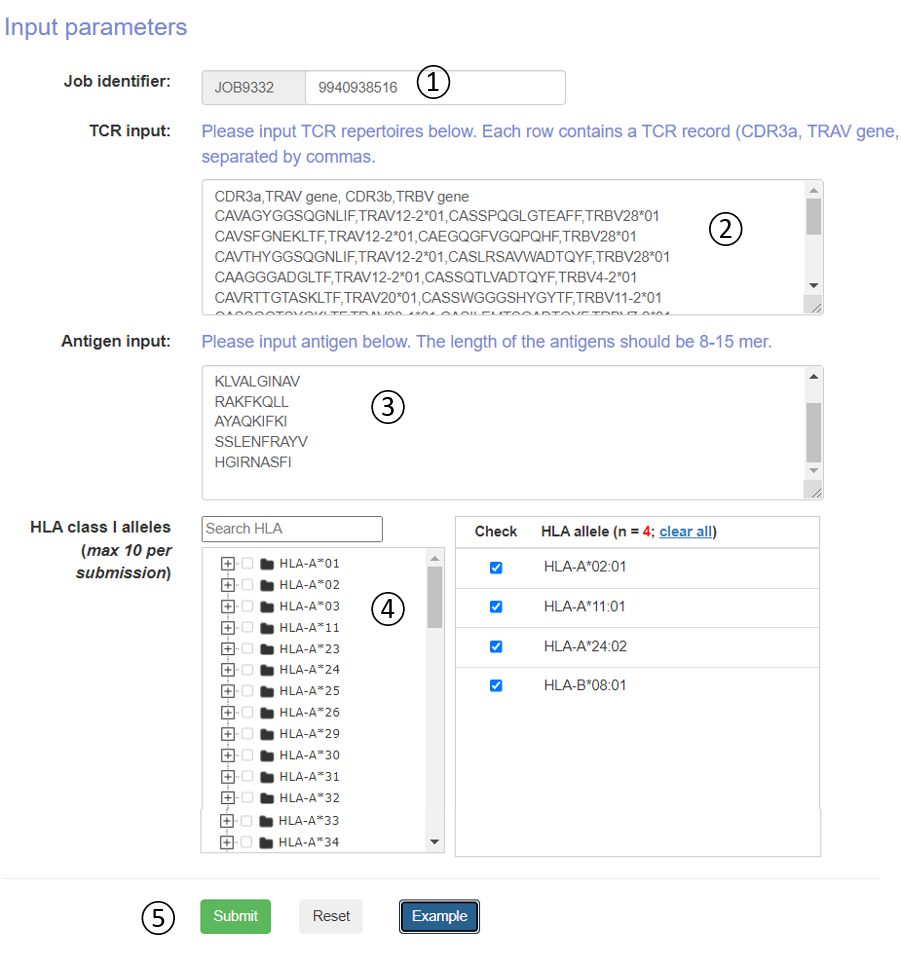
Figure 5. Job submission form.
- Job identifier: Job identifier can be generated automatically or customized by the submitter. It is confidential to other users and can be used for job status monitoring and result retrieval.(See Results page).It is required.
- TCR input: Please input TCR repertoires below. Each row contains a TCR record (CDR3a, TRAV gene, CDR3b, TRBV gene) and separated by commas.
- Antigen input: The user can directly copy the antigens in the input box. The length of the antigens should be 8-15 mer.
- HLA alleles: The DeepTCR 1.0 server predicts pMHC-TCR binding to more than >100 well studied human MHC molecule. We constructed a classification tree of HLA. Users can quickly retrieve and submit candidate HLA alleles through the search box and tree map. Each submitted task is allowed to select up to 10 HLA alleles.
- Operation buttons: Submit, reset the submission form, or access the example dataset.
Output
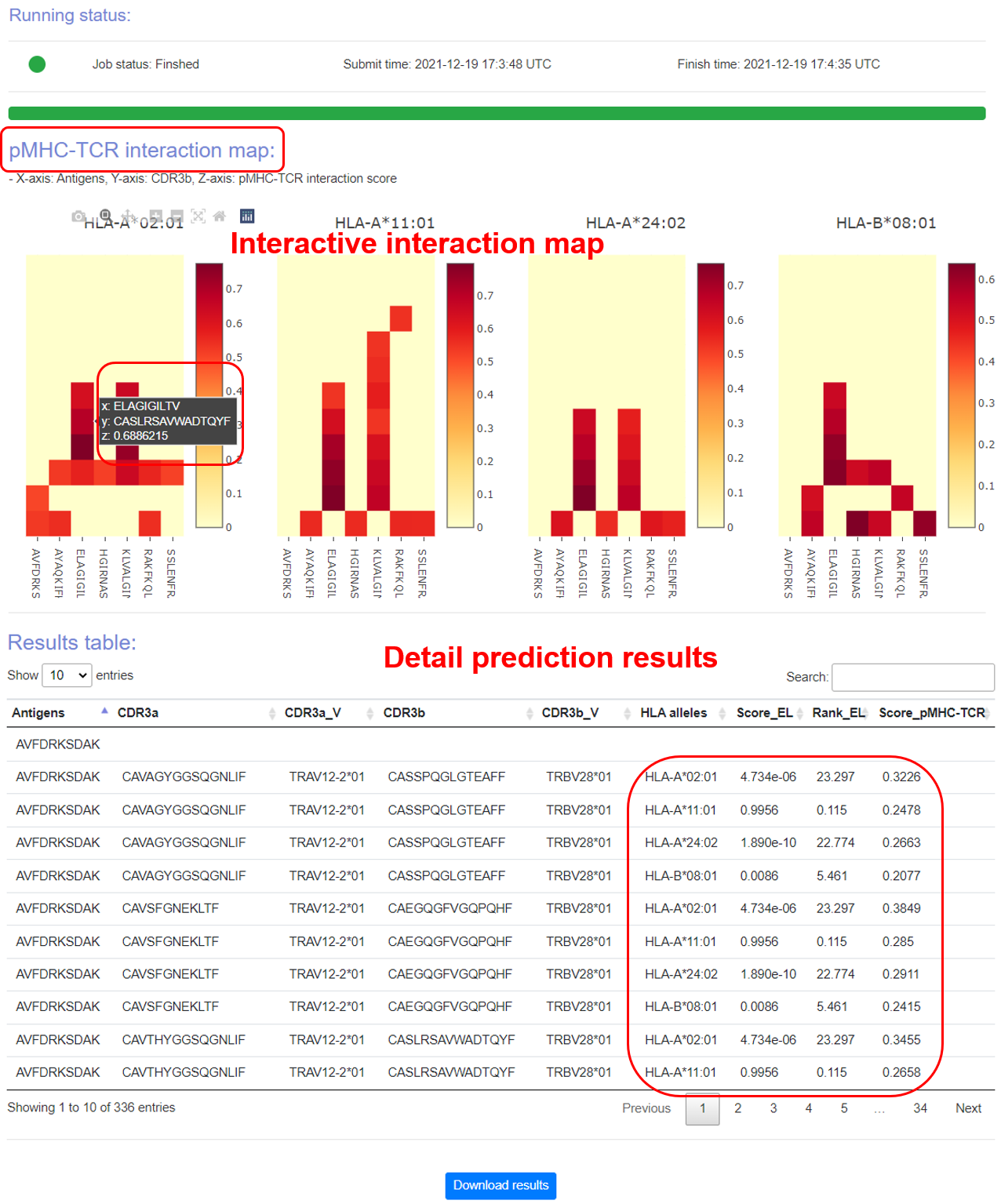
Figure 6. The prediction output.
6. Citation:
Please cite: Probing T cell response by improved TCR repertoires featurization with deep generative model. Web site: https://bioinfo.uth.edu/DeepTR.