|
|
||
|
|
||
|
As summarized in Figure 1 below, the NGS-based CNV detection methods can be categorized into five different strategies, including: (1) paired-end mapping (PEM), (2) split read (SR), (3) read depth (RD), (4) de novo assembly of a genome (AS), and (5) a combination of the above approaches (CB) .
|
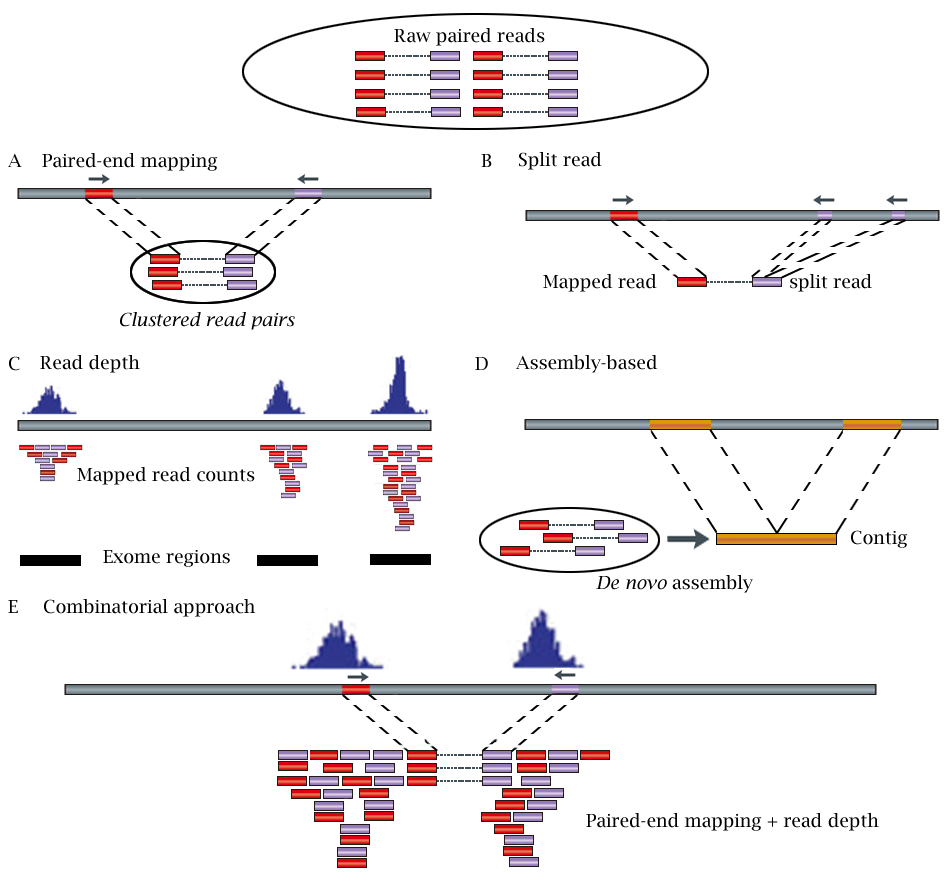
Table 1 - Summary of paired-end mapping (PEM), split read (SR), and de novo assembly (AS)-based tools for CNV detection using NGS data
Method |
URL |
Language |
Input |
Comments |
Ref. |
PEM-based |
|||||
BreakDancer |
http://breakdancer.sourceforge.net/ |
Perl, C++ |
Alignment files |
Predicting insertions, deletions, inversions, inter- and intra-chromosomal translocations |
[1] |
PEMer |
http://sv.gersteinlab.org/pemer/ |
Perl, Python |
FASTA |
Using simulation-based error models to call SVs |
[2] |
VariationHunter |
http://compbio.cs.sfu.ca/strvar.htm |
C |
DIVETa |
Detecting insertions, deletions and inversions |
[3] |
commonLAW |
http://compbio.cs.sfu.ca/strvar.htm |
C++ |
Alignment files |
Aligning multiple samples simultaneously to gain accurate SVs using maximum parsimony model |
[4] |
GASV |
http://code.google.com/p/gasv/ |
Java |
BAM |
A geometric approach for classification and comparison of structural variants |
[5] |
Spanner |
N/A |
N/A |
N/A |
Using PEM to detect tandem duplications |
[6] |
SR-based |
|||||
AGE |
http://sv.gersteinlab.org/age |
C++ |
FASTA |
A dynamic-programming algorithm using optimal alignments with gap excision to detect breakpoints |
[7] |
Pindel |
http://www.ebi.ac.uk/~kye/pindel/ |
C++ |
BAM / FASTQ |
Using a pattern growth approach to identify breakpoints of various SVs |
[8] |
SLOPE |
http://www-genepi.med.utah.edu/suppl/SLOPE |
C++ |
SAM/ FASTQ/ MAQb |
Locating SVs from targeted sequencing data |
[9] |
SRiC |
N/A |
N/A |
BLAT output |
CalibratingSV calling using realistic error models |
[10] |
AS-based |
|||||
Magnolya |
http://sourceforge.net/projects/magnolya/ |
Python |
FASTA |
Calling CNV from co-assembled genomes and estimating copy number with Poisson mixture model |
[11] |
Cortex assembler |
http://cortexassembler.sourceforge.net/ |
C |
FASTQ / FASTA |
Using alignment of de novo assembled genome to build de Bruijn graph to detect SVs |
[12] |
TIGRA-SV |
http://gmt.genome.wustl.edu/tigra-sv/ |
C |
SV callsc + BAM |
Local assembly of SVs using the iterative graph routing assembly (TIGRA) algorithm |
N/A |
|
aThe specific input format for VariationHunter, including the reads with multiple alignments. bFile format from MAQ mapview. cThe file including the detected structure variations using other tools. |
Table 2 - Read depth (RD)-based tools for CNV detection using whole genome sequencing data
Tool |
URL |
Language |
Input |
Comments |
Ref. |
SegSeqa |
http://www.broadinstitute.org/cgi-bin/cancer/publications/pub_paper.cgi?mode=view&paper_id=182 |
Matlab |
Aligned read positions |
Detecting CNV breakpoints using massively parallel sequence data |
[13] |
CNV-seqa |
http://tiger.dbs.nus.edu.sg/cnv-seq/ |
Perl, R |
Aligned read positions |
Identifying CNVs using the difference of observed copy number ratios |
[14] |
RDXplorerb |
http://rdxplorer.sourceforge.net/ |
Python, Shell |
BAM |
Detecting CNVs through event-wise testing algorithm on normalized read depth of coverage |
[15] |
BIC-seqa |
http://compbio.med.harvard.edu/Supplements/PNAS11.html |
Perl, R |
BAM |
Using the Bayesian information criterion to detect CNVs based on uniquely mapped reads |
[16] |
CNAsega |
http://www.compbio.group.cam.ac.uk/software/cnaseg |
R |
BAM |
Using flowcell-to-flowcell variability in cancer and control samples to reduce false positives |
[17] |
cn.MOPSb |
http://www.bioinf.jku.at/software/cnmops/ |
R |
BAM/read count matrices |
Modelling of read depths across samples at each genomic position using mixture Poisson model |
[18] |
JointSLMb |
http://nar.oxfordjournals.org/content/suppl/2011/02/16/ |
R |
SAM/BAM |
Population-based approach to detect common CNVs using read depth data |
[19] |
ReadDepth |
http://code.google.com/p/readdepth/ |
R |
BED files |
Using breakpoints to increase the resolution of CNV detection from low-coverage reads |
[20] |
rSW-seqa |
http://compbio.med.harvard.edu/Supplements/BMCBioinfo10-2.html |
C |
Aligned read positions |
Identifying CNVs by comparing matched tumor and control sample |
[21] |
CNVnator |
http://sv.gersteinlab.org/ |
C++ |
BAM |
Using mean-shift approach and performing multiple-bandwidth partitioning and GC correction |
[22] |
CNVnorma |
http://www.precancer.leeds.ac.uk/cnanorm |
R |
Aligned read positions |
Identifying contamination level with normal cells |
[23] |
CMDSb |
https://dsgweb.wustl.edu/qunyuan/software/cmds |
C, R |
Aligned read positions |
Discovering CNVs from multiple samples |
[24] |
mrCaNaVar |
http://mrcanavar.sourceforge.net/ |
C |
SAM |
A tool to detect large segmental duplications and insertions |
[25] |
CNVeM |
N/A |
N/A |
N/A |
Predicting CNV breakpoints in base-pair resolution |
[26] |
cnvHMM |
http://genome.wustl.edu/software/cnvhmm |
C |
consensus sequence from SAMtools |
Using HMM to detect CNV |
N/A |
|
aTools require matched case-control sample as input. bTools use multiple samples as input. |
Table 3 - Summary of bioinformatics tools for CNV detection using exome sequencing data
Tool |
URL |
Language |
Input |
Comments |
Ref. |
Control-FREECa |
http://bioinfo-out.curie.fr/projects/freec/ |
C++ |
SAM/BAM/pileup/ Eland, BED, SOAP, arachne, psi (BLAT) and Bowtie formats |
Correcting copy number using matched case-control samples or GC contents |
[27] |
CoNIFERb |
http://conifer.sf.net/ |
Python |
BAM |
Using singular value decomposition to normalize copy number and avoiding batch bias by integrating multiple samples |
[28] |
XHMMb |
http://atgu.mgh.harvard.edu/xhmm/ |
C++ |
BAM |
Uses principal component analysis to normalize copy number and HMM to detect CNVs |
[29] |
ExomeCNVc |
http://cran.r-project.org/web/packages/ExomeCNV |
R |
BAM/pileup |
Using read depth and B-allele frequencies from exome sequencing data to detect CNVs and LOHs |
[30] |
CONTRAc |
http://contra-cnv.sourceforge.net/ |
Python |
SAM/BAM |
Comparing base-level log-ratios calculated from read depth between case and control samples |
[31] |
CONDEX |
http://code.google.com/p/condr/ |
Java |
Sorted BED files |
Using HMM to identify CNVs |
[32] |
SeqGene |
http://seqgene.sourceforge.net |
Python, R |
SAM/pileup |
Calling variants, including CNVs, from exome sequencing data |
[33] |
PropSeqc |
http://bioinformatics.nki.nl/ocs/ |
R, C |
N/A |
Using the read depth of the case sample as a linear function of that of control sample to detect CNVs |
[34] |
VarScan2c |
Java |
BAM/pileup |
Using pairwise comparisons of the normalized read depth at each position to estimate CNV |
[35] |
|
ExoCNVTestb |
http://www1.imperial.ac.uk/medicine/people/l.coin/ |
Java, R |
BAM |
Identifying and genotyping common CNVs associated with complex disease |
[36] |
ExomeDepthb |
http://cran.r-project.org/web/packages/ExomeDepth/index.html |
R |
BAM |
Using beta-binomial model to fit read depth of WES data |
[37] |
|
aControl-FREEC accepts either matched case-control samples or single sample as input. bTools use multiple samples as input. cTools require matched case-control samples as input. |
Table 4 - Combinatorial bioinformatics tools for CNV detection using NGS data
Method |
URL |
Language |
Input |
Combinationa |
Ref. |
|
NovelSeq |
http://compbio.cs.sfu.ca/strvar.htm |
C |
FASTA/SAM |
PEM+AS |
[38] |
|
HYDRA |
http://code.google.com/p/hydra-sv/ |
Python |
discordant paired-end mappings |
PEM+AS |
[39] |
|
CNVer |
http://compbio.cs.toronto.edu/CNVer/ |
Perl, C++ |
BAM/ aligned positions |
PEM+RD |
[40] |
|
GASVPro |
http://code.google.com/p/gasv/ |
C++ |
BAM |
PEM+RD |
[41] |
|
Genome STRiP |
http://www.broadinstitute.org/software/ |
Java, R |
BAM |
PEM+RD |
[42] |
|
SVDetect |
http://svdetect.sourceforge.net/ |
Perl |
SAM/BAM/ ELAND |
PEM+RD |
[43] |
|
inGAP-sv |
http://ingap.sourceforge.net/ |
Java |
SAM |
PEM+RD |
[44] |
|
SVseq |
http://www.engr.uconn.edu/~jiz08001/svseq.html |
C |
FASTQ / BAM |
PEM+SR |
[45] |
|
Nord et al. |
N/A |
N/A |
N/A |
RD+SR |
[46] |
|
|
aRD: read depth-based approach; PEM: paired-end mapping approach; SR: split read approach; AS: de novo assembly approach. |
|
Reference 1. Chen K, Wallis JW, McLellan MD, Larson DE, Kalicki JM, Pohl CS, McGrath SD, Wendl MC, Zhang QY, Locke DP, et al: BreakDancer: an algorithm for high-resolution mapping of genomic structural variation. Nat Methods 2009, 6:677- 681. 2. Korbel JO, Abyzov A, Mu XJ, Carriero N, Cayting P, Zhang ZD, Snyder M, Gerstein MB: PEMer: a computational framework with simulation-based error models for inferring genomic structural variants from massive paired-end sequencing data. Genome Biol 2009, 10:R23. 3. Hormozdiari F, Hajirasouliha I, Dao P, Hach F, Yorukoglu D, Alkan C, Eichler EE, Sahinalp SC: Next-generation VariationHunter: combinatorial algorithms for transposon insertion discovery. Bioinformatics 2010, 26:i350-357. 4. Hormozdiari F, Hajirasouliha I, McPherson A, Eichler EE, Sahinalp SC: Simultaneous structural variation discovery among multiple paired-end sequenced genomes. Genome Res 2011, 21:2203-2212. 5. Sindi S, Helman E, Bashir A, Raphael BJ: A geometric approach for classification and comparison of structural variants. Bioinformatics 2009, 25:i222-230. 6. Mills RE, Walter K, Stewart C, Handsaker RE, Chen K, Alkan C, Abyzov A, Yoon SC, Ye K, Cheetham RK, et al: Mapping copy number variation by population-scale genome sequencing. Nature 2011, 470:59-65. 7. Abyzov A, Gerstein M: AGE: defining breakpoints of genomic structural variants at single-nucleotide resolution, through optimal alignments with gap excision. Bioinformatics 2011, 27:595-603. 8. Ye K, Schulz MH, Long Q, Apweiler R, Ning Z: Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics 2009, 25:2865-2871. 9. Abel HJ, Duncavage EJ, Becker N, Armstrong JR, Magrini VJ, Pfeifer JD: SLOPE: a quick and accurate method for locating non-SNP structural variation from targeted next-generation sequence data. Bioinformatics 2010, 26:2684-2688. 10. Zhang ZD, Du J, Lam H, Abyzov A, Urban AE, Snyder M, Gerstein M: Identification of genomic indels and structural variations using split reads. BMC Genomics 2011, 12:375. 11. Nijkamp JF, van den Broek MA, Geertman JM, Reinders MJ, Daran JM, de Ridder D: De novo detection of copy number variation by co-assembly. Bioinformatics 2012. 12. Iqbal Z, Caccamo M, Turner I, Flicek P, McVean G: De novo assembly and genotyping of variants using colored de Bruijn graphs. Nat Genet 2012, 44:226-232. 13. Chiang DY, Getz G, Jaffe DB, O'Kelly MJ, Zhao X, Carter SL, Russ C, Nusbaum C, Meyerson M, Lander ES: High-resolution mapping of copy-number alterations with massively parallel sequencing. Nat Methods 2009, 6:99-103. 14. Xie C, Tammi MT: CNV-seq, a new method to detect copy number variation using high-throughput sequencing. BMC Bioinformatics 2009, 10:80. 15. Yoon S, Xuan Z, Makarov V, Ye K, Sebat J: Sensitive and accurate detection of copy number variants using read depth of coverage. Genome Res 2009, 19:1586-1592. 16. Xi R, Hadjipanayis AG, Luquette LJ, Kim TM, Lee E, Zhang J, Johnson MD, Muzny DM, Wheeler DA, Gibbs RA, et al: Copy number variation detection in whole-genome sequencing data using the Bayesian information criterion. Proc Natl Acad Sci U S A 2011, 108:E1128-1136. 17. Ivakhno S, Royce T, Cox AJ, Evers DJ, Cheetham RK, Tavare S: CNAseg--a novel framework for identification of copy number changes in cancer from second-generation sequencing data. Bioinformatics 2010, 26:3051-3058. 18. Klambauer G, Schwarzbauer K, Mayr A, Clevert DA, Mitterecker A, Bodenhofer U, Hochreiter S: cn.MOPS: mixture of Poissons for discovering copy number variations in next-generation sequencing data with a low false discovery rate. Nucleic Acids Res 2012, 40:e69. 19. Magi A, Benelli M, Yoon S, Roviello F, Torricelli F: Detecting common copy number variants in high-throughput sequencing data by using JointSLM algorithm. Nucleic Acids Res 2011, 39:e65. 20. Miller CA, Hampton O, Coarfa C, Milosavljevic A: ReadDepth: a parallel R package for detecting copy number alterations from short sequencing reads. PLoS One 2011, 6:e16327. 21. Kim TM, Luquette LJ, Xi R, Park PJ: rSW-seq: algorithm for detection of copy number alterations in deep sequencing data. BMC Bioinformatics 2010, 11:432. 22. Abyzov A, Urban AE, Snyder M, Gerstein M: CNVnator: an approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res 2011, 21:974-984. 23. Gusnanto A, Wood HM, Pawitan Y, Rabbitts P, Berri S: Correcting for cancer genome size and tumour cell content enables better estimation of copy number alterations from next-generation sequence data. Bioinformatics 2012, 28:40-47. 24. Zhang Q, Ding L, Larson DE, Koboldt DC, McLellan MD, Chen K, Shi X, Kraja A, Mardis ER, Wilson RK, et al: CMDS: a population-based method for identifying recurrent DNA copy number aberrations in cancer from high-resolution data. Bioinformatics 2010, 26:464-469. 25. Alkan C, Kidd JM, Marques-Bonet T, Aksay G, Antonacci F, Hormozdiari F, Kitzman JO, Baker C, Malig M, Mutlu O, et al: Personalized copy number and segmental duplication maps using next-generation sequencing. Nat Genet 2009, 41:1061-1067. 26. Wang Z, Hormozdiari F, Yang W-Y, Halperin E, Eskin E: CNVeM: Copy Number Variation Detection Using Uncertainty of Read Mapping. In Research in Computational Molecular Biology. Volume 7262. Edited by Chor B: Springer Berlin / Heidelberg; 2012: 326-340: Lecture Notes in Computer Science]. 27. Boeva V, Zinovyev A, Bleakley K, Vert JP, Janoueix-Lerosey I, Delattre O, Barillot E: Control-free calling of copy number alterations in deep-sequencing data using GC-content normalization. Bioinformatics 2011, 27:268-269. 28. Krumm N, Sudmant PH, Ko A, O'Roak BJ, Malig M, Coe BP, Quinlan AR, Nickerson DA, Eichler EE: Copy number variation detection and genotyping from exome sequence data. Genome Res 2012, 22:1525-1532. 29. Fromer M, Moran JL, Chambert K, Banks E, Bergen SE, Ruderfer DM, Handsaker RE, McCarroll SA, O'Donovan MC, Owen MJ, et al: Discovery and Statistical Genotyping of Copy-Number Variation from Whole-Exome Sequencing Depth. Am J Hum Genet 2012, 91:597-607. 30. Sathirapongsasuti JF, Lee H, Horst BA, Brunner G, Cochran AJ, Binder S, Quackenbush J, Nelson SF: Exome sequencing-based copy-number variation and loss of heterozygosity detection: ExomeCNV. Bioinformatics 2011, 27:2648-2654. 31. Li J, Lupat R, Amarasinghe KC, Thompson ER, Doyle MA, Ryland GL, Tothill RW, Halgamuge SK, Campbell IG, Gorringe KL: CONTRA: copy number analysis for targeted resequencing. Bioinformatics 2012, 28:1307-1313. 32. Ramachandran A, Micsinai M, Pe'er I: CONDEX: Copy number detection in exome sequences. In Bioinformatics and Biomedicine Workshops (BIBMW), 2011 IEEE International Conference on; 12-15 Nov. 2011. 2011: 87-93. 33. Deng X: SeqGene: a comprehensive software solution for mining exome- and transcriptome- sequencing data. BMC Bioinformatics 2011, 12:267. 34. Rigaill GJ, Cadot S, Kluin RJ, Xue Z, Bernards R, Majewski IJ, Wessels LF: A regression model for estimating DNA copy number applied to capture sequencing data. Bioinformatics 2012, 28:2357-2365. 35. Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L, Wilson RK: VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res 2012, 22:568-576. 36. Coin LJ, Cao D, Ren J, Zuo X, Sun L, Yang S, Zhang X, Cui Y, Li Y, Jin X, Wang J: An exome sequencing pipeline for identifying and genotyping common CNVs associated with disease with application to psoriasis. Bioinformatics 2012, 28:i370-i374. 37. Plagnol V, Curtis J, Epstein M, Mok KY, Stebbings E, Grigoriadou S, Wood NW, Hambleton S, Burns SO, Thrasher AJ, et al: A robust model for read count data in exome sequencing experiments and implications for copy number variant calling. Bioinformatics 2012, 28:2747-2754. 38. Hajirasouliha I, Hormozdiari F, Alkan C, Kidd JM, Birol I, Eichler EE, Sahinalp SC: Detection and characterization of novel sequence insertions using paired-end next-generation sequencing. Bioinformatics 2010, 26:1277-1283. 39. Quinlan AR, Clark RA, Sokolova S, Leibowitz ML, Zhang Y, Hurles ME, Mell JC, Hall IM: Genome-wide mapping and assembly of structural variant breakpoints in the mouse genome. Genome Res 2010, 20:623-635. 40. Medvedev P, Fiume M, Dzamba M, Smith T, Brudno M: Detecting copy number variation with mated short reads. Genome Res 2010, 20:1613-1622. 41. Sindi SS, Onal S, Peng LC, Wu HT, Raphael BJ: An integrative probabilistic model for identification of structural variation in sequencing data. Genome Biol 2012, 13:R22. 42. Handsaker RE, Korn JM, Nemesh J, McCarroll SA: Discovery and genotyping of genome structural polymorphism by sequencing on a population scale. Nat Genet 2011, 43:269-276. 43. Zeitouni B, Boeva V, Janoueix-Lerosey I, Loeillet S, Legoix-ne P, Nicolas A, Delattre O, Barillot E: SVDetect: a tool to identify genomic structural variations from paired-end and mate-pair sequencing data. Bioinformatics 2010, 26:1895-1896. 44. Qi J, Zhao F: inGAP-sv: a novel scheme to identify and visualize structural variation from paired end mapping data. Nucleic Acids Res 2011, 39:W567-575. 45. Zhang J, Wu Y: SVseq: an approach for detecting exact breakpoints of deletions with low-coverage sequence data. Bioinformatics 2011, 27:3228-3234. 46. Nord AS, Lee M, King MC, Walsh T: Accurate and exact CNV identification from targeted high-throughput sequence data. BMC Genomics 2011, 12:184. All the above contents are revised from "Min Zhao, Qingguo Wang, Quan Wang, Peilin Jia, Zhongming Zhao: Computational tools for copy number variation (CNV) detection using next-generation sequencing data: features and perspectives. BMC Bioinformatics. Accepted". |