What is pulmonary arterial hypertension
Pulmonary hypertension
(PH) is the inappropriate elevation of pressure in the pulmonary vascular
system. PH is diagnosed by cardiac catheterization, and defined as an increase
in mean pulmonary arterial pressure of 25 mmHg or higher at rest. PH can be a
difficult diagnosis to make, as it may present at any age with symptoms that
are not uncommonly non-specific in nature. While the most common symptom is
shortness of breath with activity, it may also present with syncope, chest
pain, palpitations, and other non-specific signs and symptoms. Unfortunately,
PH is a potentially lethal disease left untreated, and often lethal despite
aggressive therapy. While most frequently associated with lung or heart
diseases, it may also be associated with a diverse group of other diseases such
as connective tissue diseases.
Pulmonary arterial
hypertension (PAH) is one type of PH. PAH is a progressive form of pulmonary
hypertension characterized by pulmonary vascular remodeling of the distal
pulmonary vasculature, ultimately leading to destruction and loss of the
smallest pulmonary arteries (1). The
ensuing syndrome of PAH is clinically characterized by reduced pulmonary arterial
circulatory flow, resulting in increased pulmonary vascular resistance, which
ultimately results in failure of the right heart (2).
In both children and adults, PAH presents as a primary disease or in
association with a diverse range of diseases such as connective tissue
diseases, portal hypertension and congenital heart disease (3). Nearly
all forms of World Health Organization Group 1 PAH demonstrate a skewed gender
ratio with significantly more females diagnosed with PAH than males (4-6).
A conservative estimate of the prevalence of isolated PAH
is 15 cases per million which represents more than 4800 patients in the United
States (6);
however, the true impact of PAH is more extensive because it also occurs in
association with multiple common systemic diseases, such as connective tissue
diseases (3). For
example, ~15% of all patients with a connective tissue disease develop PAH (7).
PAH has a very high annual mortality despite recent progress and a surge of
data generation with regard to the molecular understanding, such that a third
of all patients still die within 3 years of diagnosis (8, 9).
As a result, improved understanding of the genetic and molecular risk factors
in the pathogenesis of PAH represents a critical opportunity for the
development of effective treatments in the future. Because PAH represents one
subtype of a larger syndrome of pulmonary vascular disease (3),
and molecular advances in the field of PAH are often more widely applicable to
other forms of pulmonary vascular disease, progress in the PAH research field
often benefits PH understanding more broadly.
As shown in the Figure below, the pathology of PAH
involves multiple processes/factors which influence vascular remodeling. These
factors include vasoactive factor imbalance, growth factors, exuberant inflammatory
cell recruitment, endothelial cell resistance to apoptosis, and vascular smooth
muscle cell growth & proliferation.
Germline bone morphogenetic protein receptor type 2 (BMPR2)
gene mutations are responsible for HPAH in 80-85% of families with a family
history of PAH, while 5-25% of patients diagnosed as having idiopathic PAH (IPAH)
actually have a detectable germline mutation in BMPR2 as well (10-16). BMPR2 mutations constitute the largest known risk for developing PAH. However, recent
studies have uncovered additional rare and common variants relevant to disease pathogenesis. However, subjects who are BMPR2 mutation carriers have only a 26% chance of developing disease (17). Thus, the penetrance of BMPR2-associated PAH is reduced, which suggests
that as yet unknown factors modify disease risk by influencing normal
homeostatic mechanisms in the pulmonary vasculature (18).
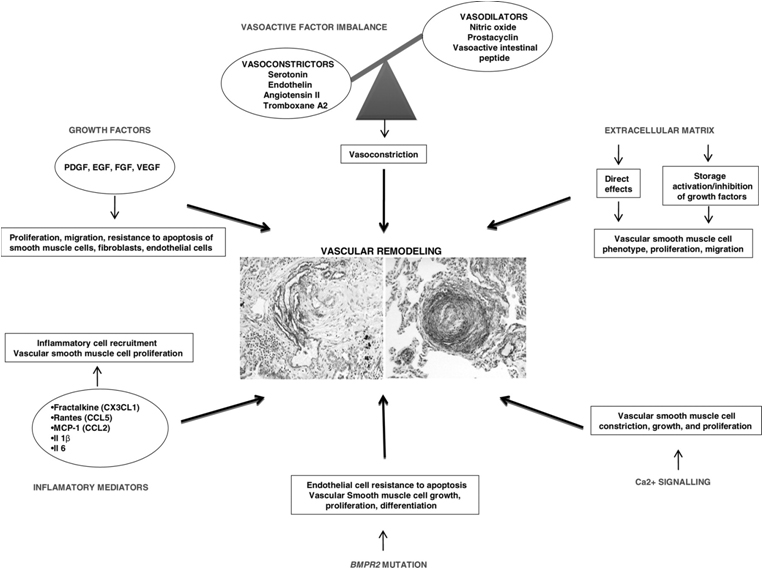
Adopted from Toshner M et al. Br Med
Bull 2010;94:21-32 (http://bmb.oxfordjournals.org/content/94/1/21.full.pdf+html) with permission from Oxford University Press.
Acknowledgment and statement: This permission is limited to this particular use and does not
allow the user to use it elsewhere or in any other format other than in this
document page. The above OUP figure is view only and does not come under a
Creative Commons license (http://creativecommons.org/licenses/) that would allow reuse without requiring permission from OUP. For permissions, please contact [email protected].
Below is a selection of relevant
publications in the field of PAH with particular focus on the genetic features
of the disease, although there is a vast and excellent body of literature
beyond this selection.
References:
1. Tuder RM, Abman SH, Braun T, Capron F,
Stevens T, Thistlethwaite PA, Haworth SG. Development and pathology of
pulmonary hypertension. J Am Coll Cardiol 2009;54:S3-9.
2. McLaughlin VV, Archer SL, Badesch DB,
Barst RJ, Farber HW, Lindner JR, Mathier MA, McGoon MD, Park MH, Rosenson RS,
Rubin LJ, Tapson VF, Varga J. Accf/aha 2009 expert consensus document on
pulmonary hypertension a report of the american college of cardiology
foundation task force on expert consensus documents and the american heart
association developed in collaboration with the american college of chest
physicians; american thoracic society, inc.; and the pulmonary hypertension
association. J Am Coll Cardiol 2009;53:1573-1619.
3. Simonneau G, Robbins IM, Beghetti M,
Channick RN, Delcroix M, Denton CP, Elliott CG, Gaine SP, Gladwin MT, Jing ZC,
Krowka MJ, Langleben D, Nakanishi N, Souza R. Updated clinical classification
of pulmonary hypertension. J Am Coll Cardiol 2009;54:S43-54.
4. Badesch DB, Raskob GE, Elliott CG,
Krichman AM, Farber HW, Frost AE, Barst RJ, Benza RL, Liou TG, Turner M, Giles
S, Feldkircher K, Miller DP, McGoon M. Pulmonary arterial hypertension:
Baseline characteristics from the reveal registry. Chest 2009.
5. Chin KM, Rubin LJ. Pulmonary arterial
hypertension. J Am Coll Cardiol 2008;51:1527-1538.
6. Humbert M, Sitbon O, Chaouat A,
Bertocchi M, Habib G, Gressin V, Yaici A, Weitzenblum E, Cordier JF, Chabot F,
Dromer C, Pison C, Reynaud-Gaubert M, Haloun A, Laurent M, Hachulla E,
Simonneau G. Pulmonary arterial hypertension in france: Results from a national
registry. Am J Respir Crit Care Med 2006;173:1023-1030.
7. Yang X, Mardekian J, Sanders KN,
Mychaskiw MA, Thomas J, 3rd. Prevalence of pulmonary arterial hypertension in
patients with connective tissue diseases: A systematic review of the
literature. Clinical rheumatology 2013.
8. Humbert M, Sitbon O, Yaici A, Montani
D, O'Callaghan DS, Jais X, Parent F, Savale L, Natali D, Gunther S, Chaouat A,
Chabot F, Cordier JF, Habib G, Gressin V, Jing ZC, Souza R, Simonneau G.
Survival in incident and prevalent cohorts of patients with pulmonary arterial
hypertension. Eur Respir J;36:549-555.
9. Macchia A, Marchioli R, Tognoni G,
Scarano M, Marfisi R, Tavazzi L, Rich S. Systematic review of trials using
vasodilators in pulmonary arterial hypertension: Why a new approach is needed. Am
Heart J 2010;159:245-257.
10. Aldred MA, Vijayakrishnan J, James V,
Soubrier F, Gomez-Sanchez MA, Martensson G, Galie N, Manes A, Corris P,
Simonneau G, Humbert M, Morrell NW, Trembath RC. Bmpr2 gene rearrangements
account for a significant proportion of mutations in familial and idiopathic
pulmonary arterial hypertension. Hum Mutat 2006;27:212-213.
11. Fujiwara M, Yagi H, Matsuoka R,
Akimoto K, Furutani M, Imamura S, Uehara R, Nakayama T, Takao A, Nakazawa M,
Saji T. Implications of mutations of activin receptor-like kinase 1 gene (alk1)
in addition to bone morphogenetic protein receptor ii gene (bmpr2) in children
with pulmonary arterial hypertension. Circulation journal : official journal
of the Japanese Circulation Society 2008;72:127-133.
12. Lane KB, Machado RD, Pauciulo MW,
Thomson JR, Phillips JA, 3rd, Loyd JE, Nichols WC, Trembath RC. Heterozygous
germline mutations in bmpr2, encoding a tgf-beta receptor, cause familial
primary pulmonary hypertension. The international pph consortium. Nat Genet 2000;26:81-84.
13. Machado RD, Aldred MA, James V,
Harrison RE, Patel B, Schwalbe EC, Gruenig E, Janssen B, Koehler R, Seeger W,
Eickelberg O, Olschewski H, Elliott CG, Glissmeyer E, Carlquist J, Kim M,
Torbicki A, Fijalkowska A, Szewczyk G, Parma J, Abramowicz MJ, Galie N,
Morisaki H, Kyotani S, Nakanishi N, Morisaki T, Humbert M, Simonneau G, Sitbon
O, Soubrier F, Coulet F, Morrell NW, Trembath RC. Mutations of the tgf-beta
type ii receptor bmpr2 in pulmonary arterial hypertension. Hum Mutat 2006;27:121-132.
14. Thomson J, Machado R, Pauciulo M,
Morgan N, Yacoub M, Corris P, McNeil K, Loyd J, Nichols W, Trembath R. Familial
and sporadic primary pulmonary hypertension is caused by bmpr2 gene mutations
resulting in haploinsufficiency of the bone morphogenetic protein tuype ii
receptor. J Heart Lung Transplant 2001;20:149.
15. Thomson JR, Machado RD, Pauciulo MW,
Morgan NV, Humbert M, Elliott GC, Ward K, Yacoub M, Mikhail G, Rogers P, Newman
J, Wheeler L, Higenbottam T, Gibbs JSR, Egan J, Crozier A, Peacock A, Allcock
R, Corris P, Loyd JE, Trembath RC, Nichols WC. Sporadic primary pulmonary
hypertension is associated with germline mutations of the gene encoding
bmpr-ii, a receptor member of the tgf-beta family. J Med Genet 2000;37:741-745.
16. Austin ED, Phillips JA, 3rd, Cogan JD,
Hamid R, Yu C, Stanton KC, Phillips CA, Wheeler LA, Robbins IM, Newman JH, Loyd
JE. Truncating and missense bmpr2 mutations differentially affect the severity
of heritable pulmonary arterial hypertension. Respir Res 2009;10:87.
17. Larkin EK, Newman JH, Austin ED, Hemnes
AR, Wheeler L, Robbins IM, West JD, Phillips JA, 3rd, Hamid R, Loyd JE.
Longitudinal analysis casts doubt on the presence of genetic anticipation in
heritable pulmonary arterial hypertension. Am J Respir Crit Care Med 2012;186:892-896.
18.Newman
JH, Phillips JA, 3rd, Loyd JE. Narrative review: The enigma of pulmonary
arterial hypertension: New insights from genetic studies. Ann Intern Med 2008;148:278-283.
