DeepFun
DeepFun is a deep-learning-based model for functional evaluation of genetic variants at the single-base resolution. Specifically, DeepFun is a comprehensive collection of chromatin profiles from ENCODE and Roadmap projects, which constructs the feature space, including 1548 DNase I accessibility, 1536 histone mark, and 4795 transcription factor binding profiles covering 225 tissues or cell lines. With such comprehensive epi-genomics annotations and the pre-trained model, this web-server provides an online service to quickly assess the impact of genetic variants in a tissue- or cell-type-specific manner.
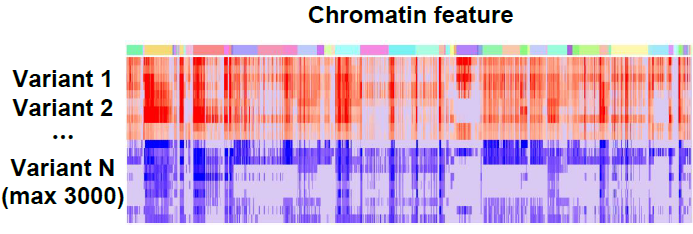
Screen analysis
For a list of query variants across all chromatin features, the screen analysis computes SNP Activity (accessibility or binding probability) Difference (SAD) or relative log fold change of odds (log-odds) difference between two alleles.
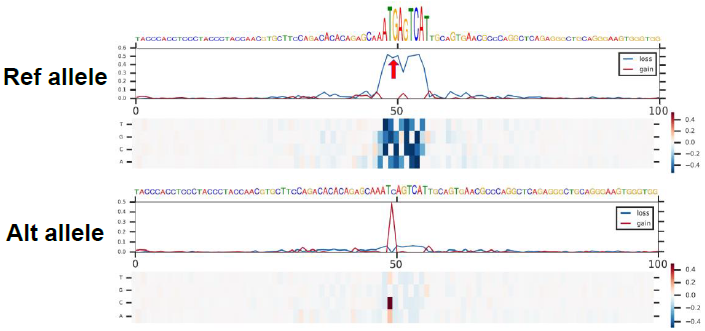
In silico saturated mutagenesis analysis
Perform an in silico saturated mutagenesis analysis of the regions surrounding 200 bp of one query variant under a target chromatin feature of interest.
Developers:
Ruifeng Hu, Guangsheng Pei, Peilin Jia, Zhongming Zhao @ CPH, UTHealth-Houston SBMI.
DeepFun is free and open to all users and there is no login requirement.
Reference:
Pei, G., Hu, R., Dai, Y., Manuel, A.M., Zhao, Z. and Jia, P. (2021) Predicting regulatory variants using a dense epigenomic mapped CNN model elucidated the molecular basis of trait-tissue associations. Nucleic Acids Res, 49, 53-66.