Tutorial
1. Basic Information
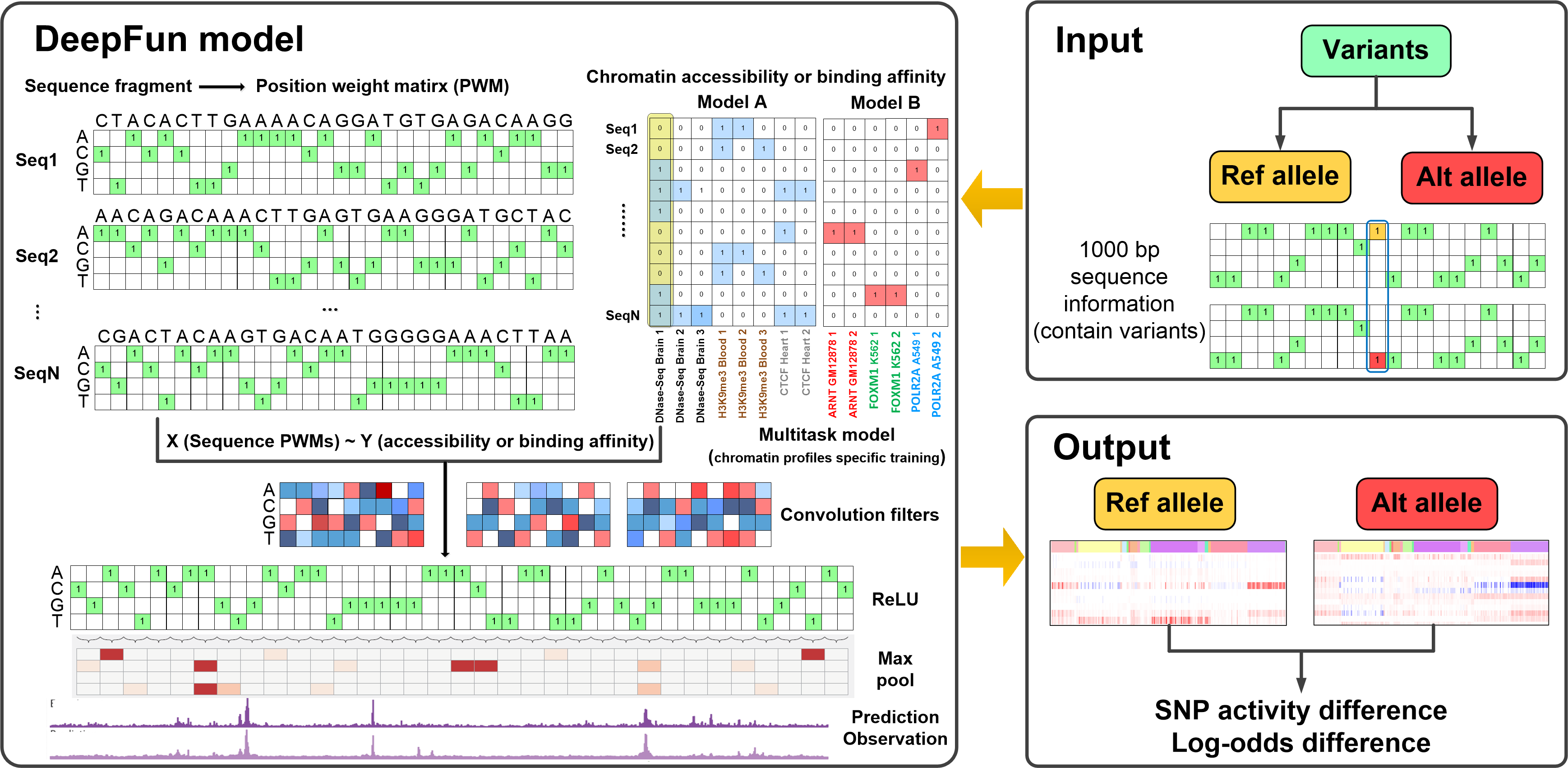
DeepFun aims to predict the impact of genetic variants on a wide range of chromatin features, based on deep convolutional neural networks (CNN) model. DeepFun provides two major functions: “screen analysis” and "in silico saturated mutagenesis" analysis.
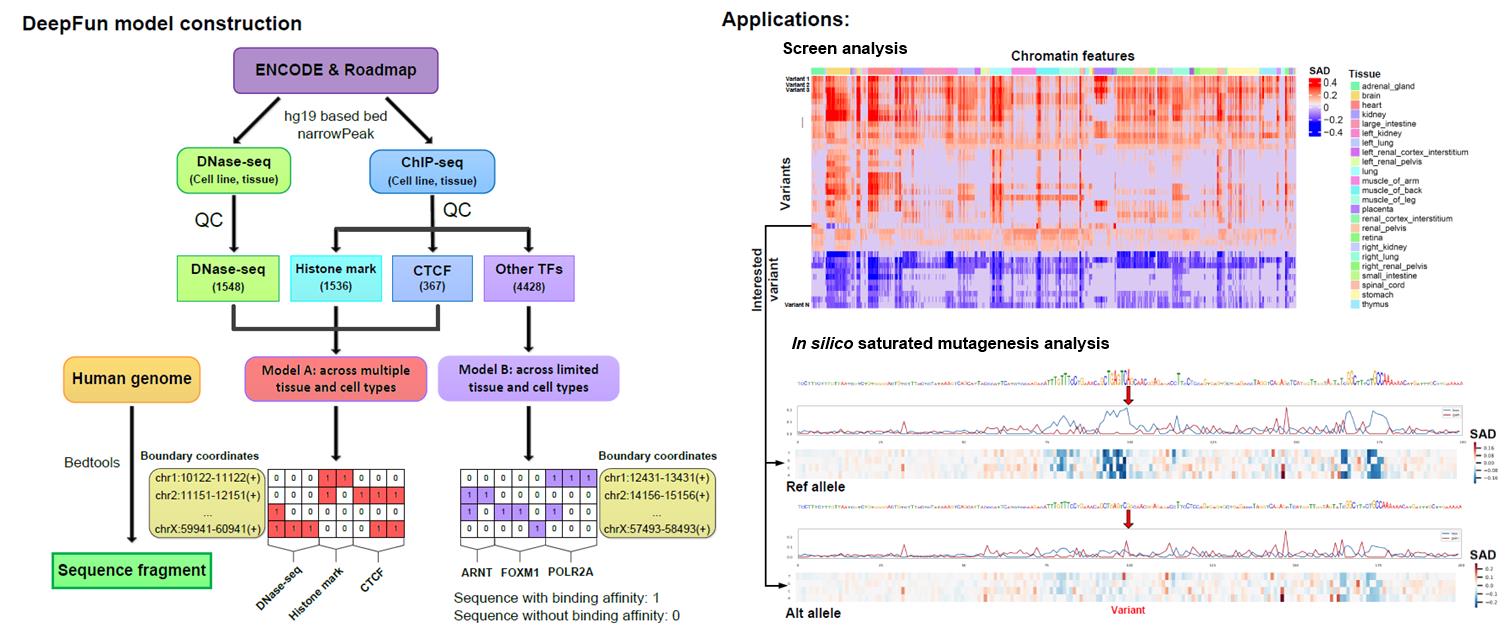
The “screen analysis” predicts the impact of a list of query variants across 7879 chromatin features (See full list here). For each variant, DeepFun considers all variants within a 1000 bp region, and use the context information of the reference and the alternative allele of each variant. Then, DeepFun predicts the activity (accessibility or binding) probability of the sequence containing the reference allele or alternative allele, based on all chromatin features in the selected model (range from 0 to 1). Two measurements, SNP activity (accessibility or binding probability) difference (SAD) and relative log fold change of odds difference between two alleles (log-odds difference) score, were implemented to evaluate the impact of the variant. It is important to note that the DeepFun model does not take any specific variant information into consideration. The screen analysis feature in DeepFun enables the prediction of variant-impacts, even for many that were never previously observed.
The “in silico saturated mutagenesis analysis” dissects the impact of one specific variant of the 200 bp region surrounding one specified chromatin feature, to discover informative sequence features. Specifically, DeepFun mutates every single base within 200 bps of the query variant and calculates the SAD change pattern in the target chromatin feature.
Note: Currently, we allow input variants based on the human genome version hg19/GRCh37 and hg38/GRCh38 (liftOver). The training of DeepFun model was based on hg19/GRCh37. Thus, for input variants based on hg38/GRCh38 coordinates, we first use liftOver to convert the coordinates to hg19/GRCh37, and then run the job. Variants that cannot be mapped to appropriate hg19/GRCh37 coordinates will be excluded from downstream analyses.
2. Model construction and evaluation
The DeepFun framework uses the expanded chromatin features resources from ENCODE Project Consortium and Roadmap Epigenomics Consortium (May 06, 2019): DNase-seq (DNA accessibility profiles) and ChIP-seq (include histone mark and transcription factor (TF) binding profiles) (See full list here). According to the assay’s functional category and completeness, we classified them into two models. Model A assays include DNase-seq, histone marks and transcription factor CTCF binding profiles across 3451 total experiments; Model B assays include remain 305 transcription factor binding profiles across 4428 total experiments.
For datasets in both models A and B, the assays are reformatted with 1000 bp genomic intervals by extending 500 bp on each side of the midpoint of any narrow peaks reported in the original dataset, according to the Basset configuration. We then greedily merged peaks based on their distance to an adjacent peak, until no peaks overlapped by >200 bp (processed by the preprocess_features.py function). The center of the merged peak was determined as a weighted average of the midpoints of the merged peaks from the individual profile. These peaks were regarded as potential epigenomic active sites. Next, we applied an extended version of the Basset model with 3 default convolutional layers, two fully connected hidden layers and a fully connected sigmoid transformation layer, to predict the peak accessibility or binding probability across different chromatin features. We randomly selected 80% of epigenomic active sites for training, 10% for validation, and 10% for testing, respectively. We used the area under receiver operating characteristic (AUC) to evaluate the performance on validation and testing sets. The network training was stopped when the loss in the validation set did not decrease within 12 successive epochs of Bayesian optimization. By this measurement, DeepFun achieved a median AUC of 0.933, 0.872 and 0.80 over DNase-seq, histone mark and transcription factor assays, respectively. Detailed description of DeepFun model construction and evaluation, and several independent validations are available from our recent work (Pei et. al 2021).
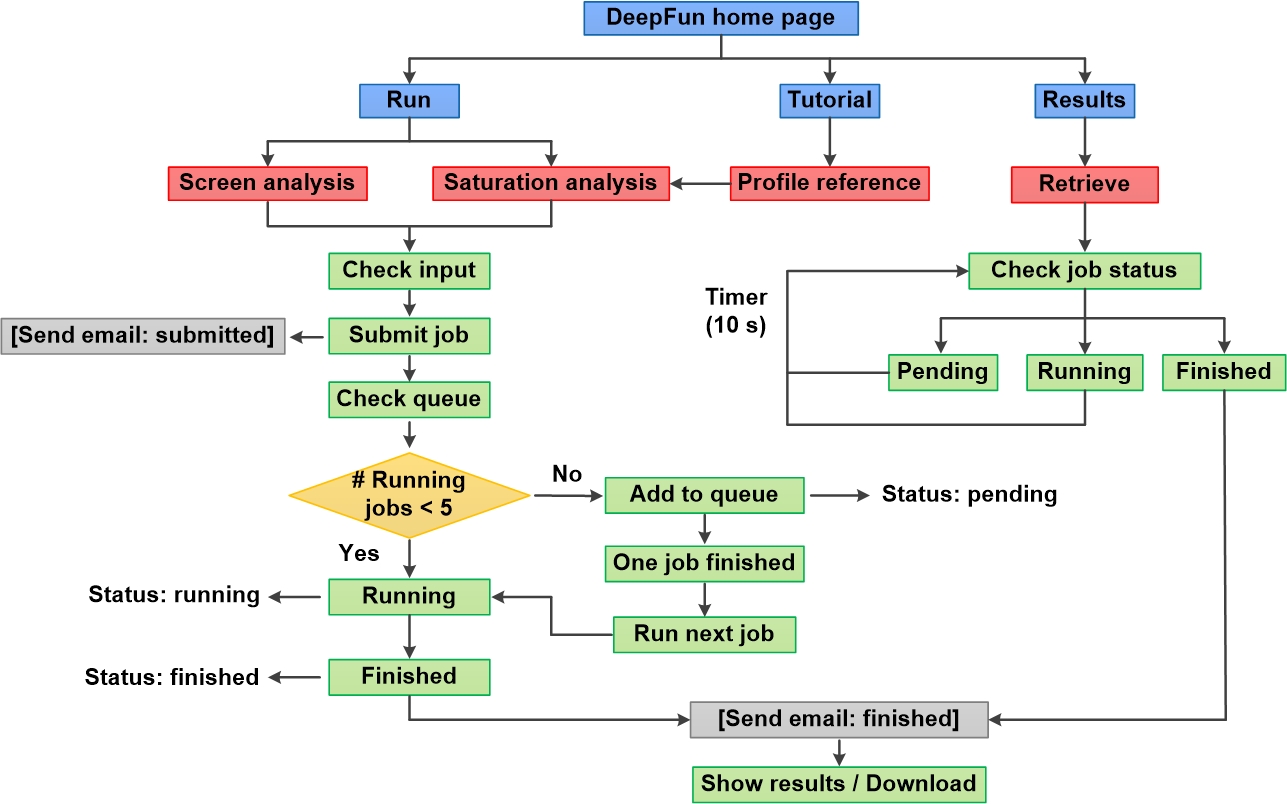
3. General workflow of DeepFun webserver
4. Screen analysis
The screen analysis calculates the SNP activity difference (SAD) and relative log fold change of odds (log-odds) difference between two alleles across all chromatin features for a number of variants (up to 3000 per job). It is mainly used for a preliminary screen, which can discover potential regulatory functional variants, e.g., ranking the query variants based on their SAD or log-odds difference. Variants with a higher positive SAD value indicate that the alternative allele increases the activity (accessibility or binding) signal compared to the reference allele, while a negative value indicates a decrease in the activity (accessibility or binding) signal.
4.1 Input:
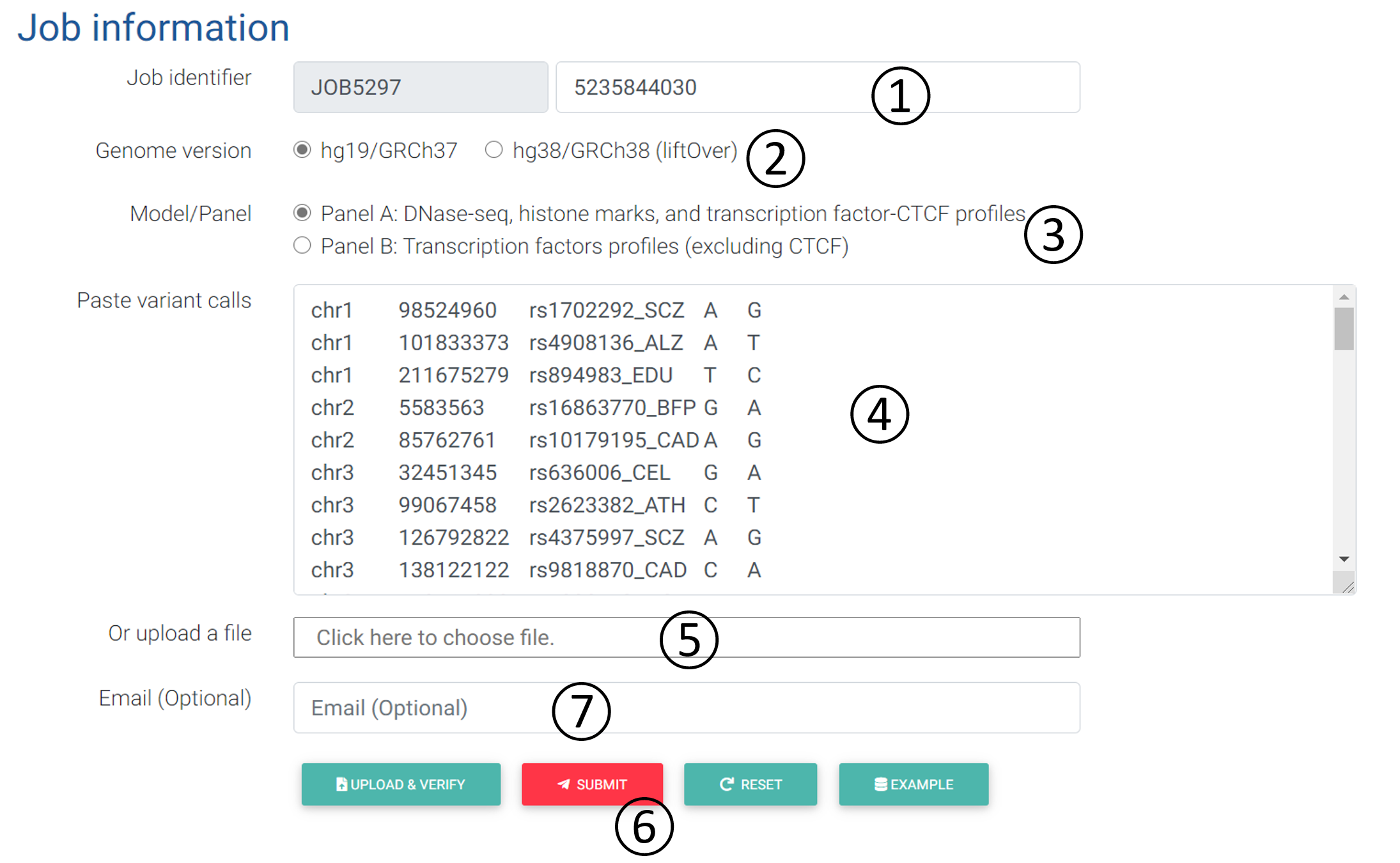
- Job Identifier: The job identifier can be generated automatically or customized by the submitter. It is confidential and required for job status monitoring and result retrieval. (See Results page).It is required.
- Genome version: DeepFun supports the coordinates from the human genome version hg19/GRCh37 (default training) or hg38/GRCh38 (liftOver).
- Model/Panel: Model A integrated 3451 profiles, including DNase-seq (1548), histone mark (1536) and transcription factor CTCF (367) profiles. Model B integrated 4428 profiles, including all remaining transcription factors binding profiles.
- Paste variant calls: Input box: The format of input can simply be 4-column text, including chromosome, position, reference allele, and alternative allele, separate by space or tab, where each variant is in one line. The VCF-like format (without header) is also accepted: 5 columns or more, including chromosome, position, SNP ID, reference allele, alternative allele, combined with other information. Each screen analysis job allows a maximum number of 3000 variants. DeepFun only accepts variant located on Chr 1 to 22 and X. Any variants located on Chr Y or mitochondria will be declined.
- Upload a file: In the screen analysis, the user can upload a file containing the variants in the required format (see above).
- Operation buttons: After inputting all the necessary information, the user should verify the inputs first. If passed, the submit button will be activated for job submission.
- Email: Users can provide their email address for receiving notifications of job status (Submitted/Finished).
4.2 Output
A downloading link will be available after the job is finished. All output files are zipped into one downloadable file. An example can be accessed HERE.

- Ref: Predicted chromatin feature activity probabilities for sequences carrying the reference allele. The row and column represent variants and all chromatin features within user selected model.
- Alt: Predicted chromatin feature activity probabilities for sequences carrying the alternative allele. The row and column represent variants and all chromatin features within user selected model.
- SAD: Predicted changes of chromatin feature activity probabilities between two alleles, defined as SAD
SAD = Alt - Ref
The row and column represent variants and all chromatin features within user selected model. - log_odds_diff: Predicted log fold change of odds (log-odds) difference between two alleles, defined as log-odds difference.
log-odds difference = log(Alt/(1-Alt)) - log(Ref/(1 - Ref))
The row and column represent variants and all chromatin features within user selected model. - SAD_summary: The SAD summary over all chromatin features and the top most significant associated chromatin features (based on SAD value). The row represent variants, the column include the SAD average, SAD standard deviation, SAD max, SAD max feature, SAD min, SAD min feature, and ANNOVAR annotation from refGene database.
- log_odds_diff_summary: The log-odds difference summary over all chromatin features and the top most significant associated chromatin feature (based on log-odds difference value). The row represent variants, the column include the log_odds difference average, log_odds difference standard deviation, log_odds difference max, log_odds difference max feature, log_odds difference min, log_odds difference min feature, and ANNOVAR annotation from refGene database.
In addition, we also present several heat maps to better demonstrate the impact of a variant in a tissue and cell type manner. It is noted that only the top 50 variants with the highest maximum SAD will be displayed.
4.3 Downstream analysis
For a better custom figure, we have deposited the codes for downstream analysis (heatmap plot, quality control, evaluate significant changes) at Github. We also deposited DeepFun pre-trained models at Github to facilitate users in running the pre-trained models on their local hardware.
5. In silico saturated mutagenesis.
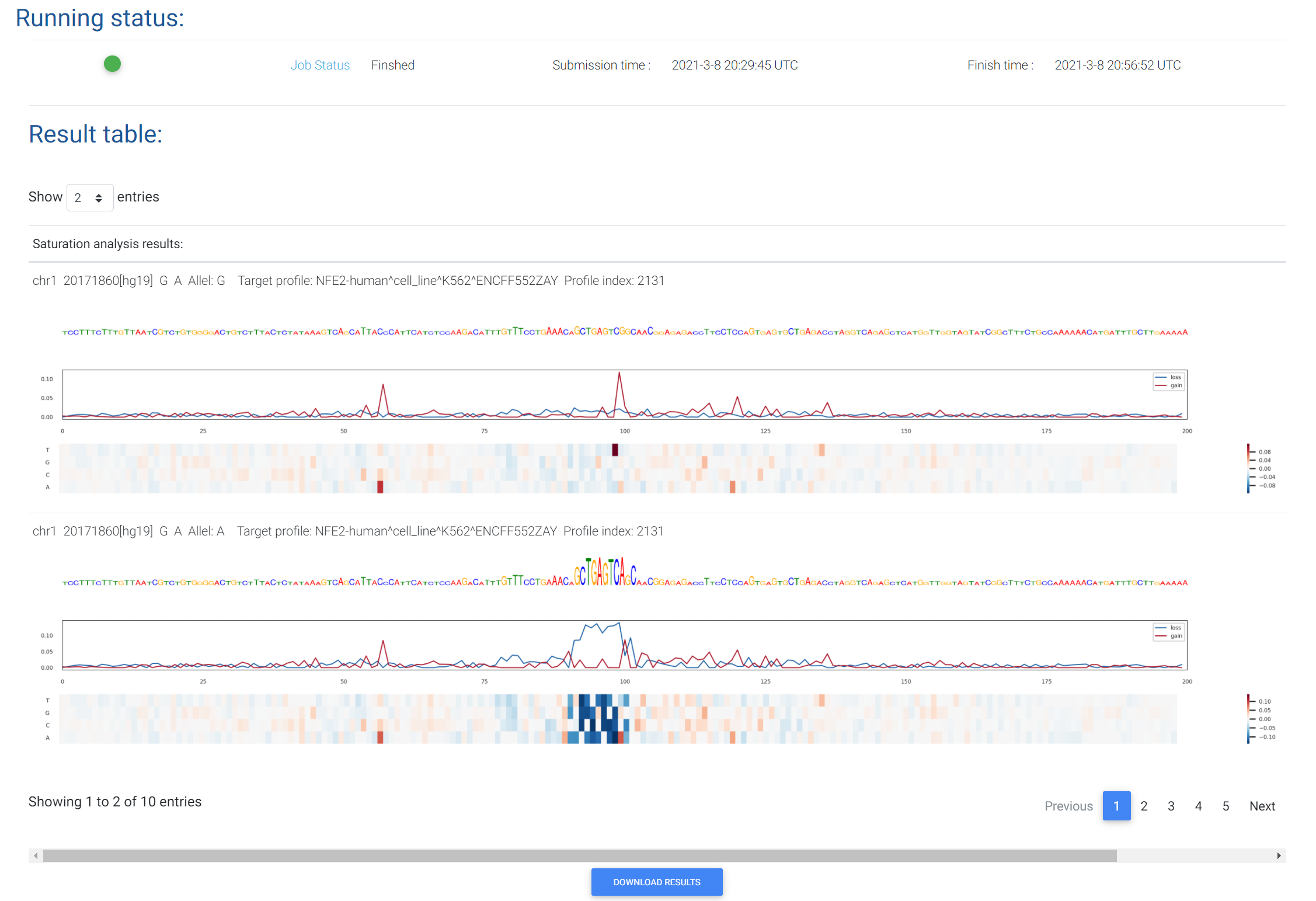
DeepFun performs "in silico saturated mutagenesis" analysis to discover potential informative sequence features. Specifically, it will mutate every single base in the input sequence, considering the reference allele and the alternative allele, and calculates the changes of sequence activity probabilities. Note that the in silico saturated mutagenesis is time-consuming. Currently, DeepFun only accepts one variant per job. Users need to specify the target chromatin feature of interest. This can be determined by the screen analysis. Typically the top 1~10 most informative features are of interest for the in silico saturated mutagenesis.
5.1 Input:
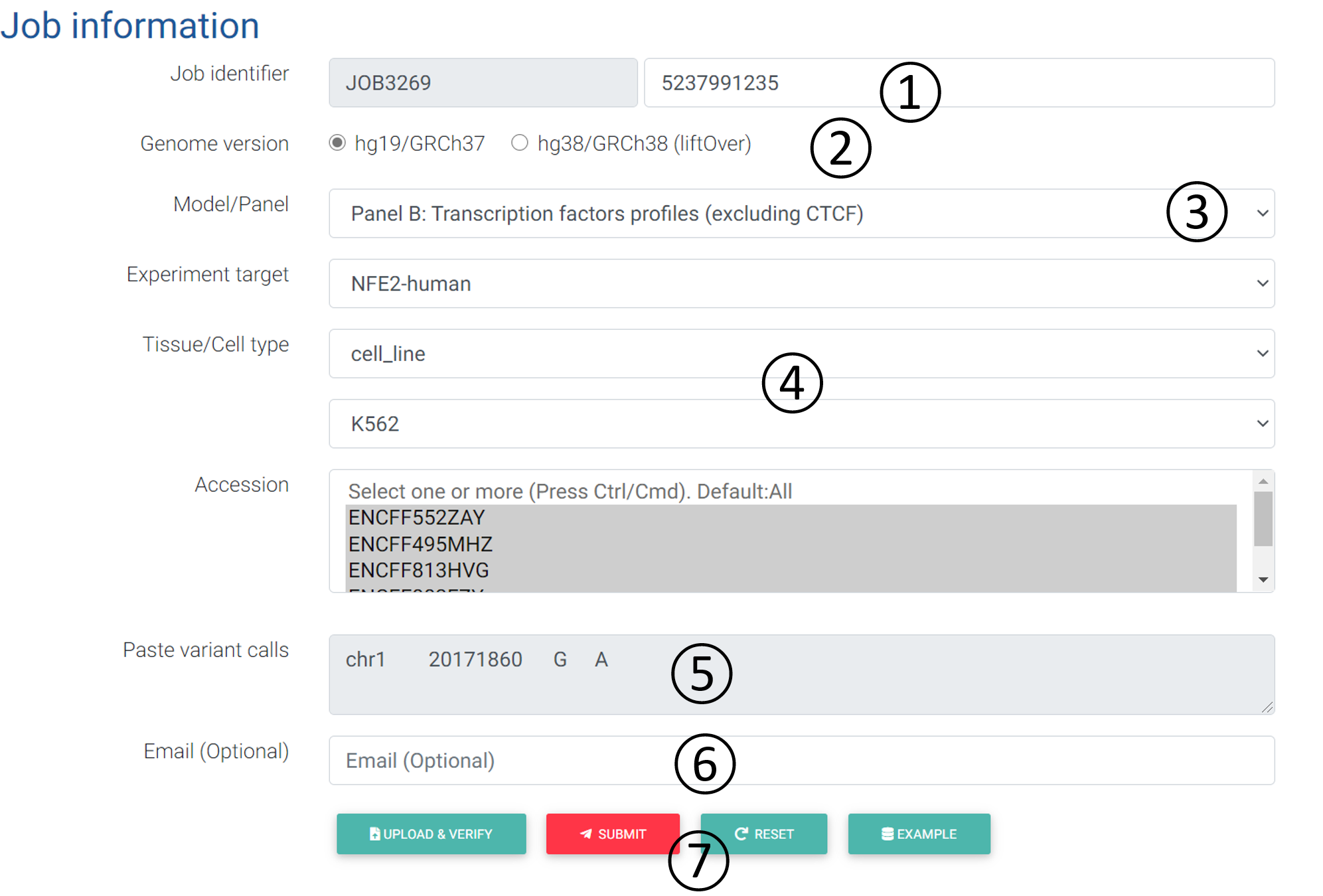
- Job Identifier: The job identifier can be generated automatically or customized by the submitter. It is confidential and required for job status monitoring and result retrieval. (See Results page).It is required.
- Genome version: DeepFun supports the coordinates from the human genome version hg19/GRCh37 (default training) or hg38/GRCh38 (liftOver).
- Model/Panel: Model A integrated 3451 profiles, including DNase-seq (1548), histone mark (1536) and transcription factor CTCF (367) profiles. Model B integrated 4428 profiles, including all remaining transcription factors binding profiles.
- Selection of user-specified profiles.
- Experiment target: (a) DNase-seq, histone marks, and transcription factor-CTCF profiles;
(b) Transcription factors profiles (excluding CTCF). - Tissue/Cell type: Available tissue or cell type with selected chromatin feature.
- Accession: replicates of chromatin features (See full profile list here).
- Experiment target: (a) DNase-seq, histone marks, and transcription factor-CTCF profiles;
- Paste variant calls: Input box: The format of input can simply be 4-column text, including chromosome, position, reference allele, and alternative allele, separate by space or tab, where each variant is in one line. The VCF-like format (without header) is also accepted: 5 columns or more, including chromosome, position, SNP ID, reference allele, alternative allele, combined with other information. Each screen analysis job allows a maximum number of 3000 variants. DeepFun only accepts variant located on Chr 1 to 22 and X. Any variants located on Chr Y or mitochondria will be declined.
- Email: Users can provide their email address for receiving notifications of job status (Submitted/Finished).
- Operation buttons: After inputting all the necessary information, the user should first verify the inputs. If passed, the submit button will be activated for job submission.
5.2 Output
For each chromatin feature, DeepFun in silico saturated mutagenesis analysis will provide two heat maps to display the SAD change patterns around the both reference and alternative alleles of variant (-99 to 100 bp). A results table containing the max gain and loss SAD value from variant’s upstream 99 to downstream 100 bp will also be provided in the results. An example can be accessed HERE.