Introduction
The selection of human leukocyte antigen (HLA) epitopes is critical for advancing cancer immunotherapy strategies and vaccine development. Recent strides in liquid chromatography and mass spectrometry have expedited the in-depth characterization of the HLA-presented ligandome. Concurrently with these technological advancements, the development of efficient methods for deciphering immunopeptidomics data and robust (neo)antigen presentation predictors is urgently needed with vast potential. Here, we developed the ImmuneApp, which facilitates prediction of antigen presentation, scoring for neoepitope immunogenicity, and immunopeptidomics analysis, with enhanced precision. ImmuneApp harnesses an interpretable, attention-based hybrid deep learning framework for predicting HLA-I epitopes trained on 349,650 ligands, enabling the extraction of informative embeddings and identification of critical residues involved in mediating pHLA binding specificity. Evaluation conducted on independent mono-allelic dataset demonstrated that ImmuneApp significantly outperforms existing methods for antigen presentation prediction. Additionally, we present a more accurate model-based deconvolution approach and conduct a systematic analysis of 216 publicly available multi-allelic immunopeptidomics samples, resulting in the deconvolution of 835,551 ligands restricted to over 100 distinct HLA-I alleles. Our investigation highlights the effectiveness of a composite model, denoted as ImmuneApp-MA, which integrates both mono- and multi-allelic data modalities to enhance predictive performance. Leveraging ImmuneApp-MA as a pre-trained model for deep transfer learning on a curated immunogenicity dataset, we introduce ImmuneApp-Neo, a novel immunogenicity predictor that outperforms existing state-of-the-art methods in prioritizing immunogenic neoepitopes, yielding a notable 2.1-fold improvement in positive predictive value (PPV). We further demonstrate the utility of ImmuneApp across diverse disease-related immunopeptidomics datasets sourced from tumor tissues and cancer biopsies, highlighting its efficacy in various tasks including quality control, binding annotations, HLA assignment, motif discovery and elucidation, and antigen presentation prediction on a sample-specific basis.
ImmuneApp workflow
By incorporating a large number of in vitro binding measurements and the ligand data of Mass spectrometry (MS)-based immunopeptidomics, we first constructed a new pan-allele MHC class I predictor through the CNN-LSTM neural network. On this basis, we further developed a semi-automated online tool called ImmuneApp that enables rapid evaluation and analysis of multiple MHC class I immunopeptidomic datasets. ImmuneApp mainly provide two services: 1. Clinical immunopeptidomic cohorts analysis of tumor biopsies; 2. Antigen presentation prediction tasks.
1. Immunopeptidomic cohorts analysis:
- Quality control
- Globle binding map
- HLA assignment
- Motif discovery (unsupervised and supervised)
- Decomposition
- Antigen presentation prediction
2. Antigen presentation prediction:
- Transfer learning enhanced neoepitopes screen
- Eluted ligand likelihood prediction
- Overall antigen presentation prediction
3. neoepitopes immunogenicity screening:
- In vitro binding measurements prediction
- Prioritizing immunogenic neoepitopes
- Greater PPV compared to all other models
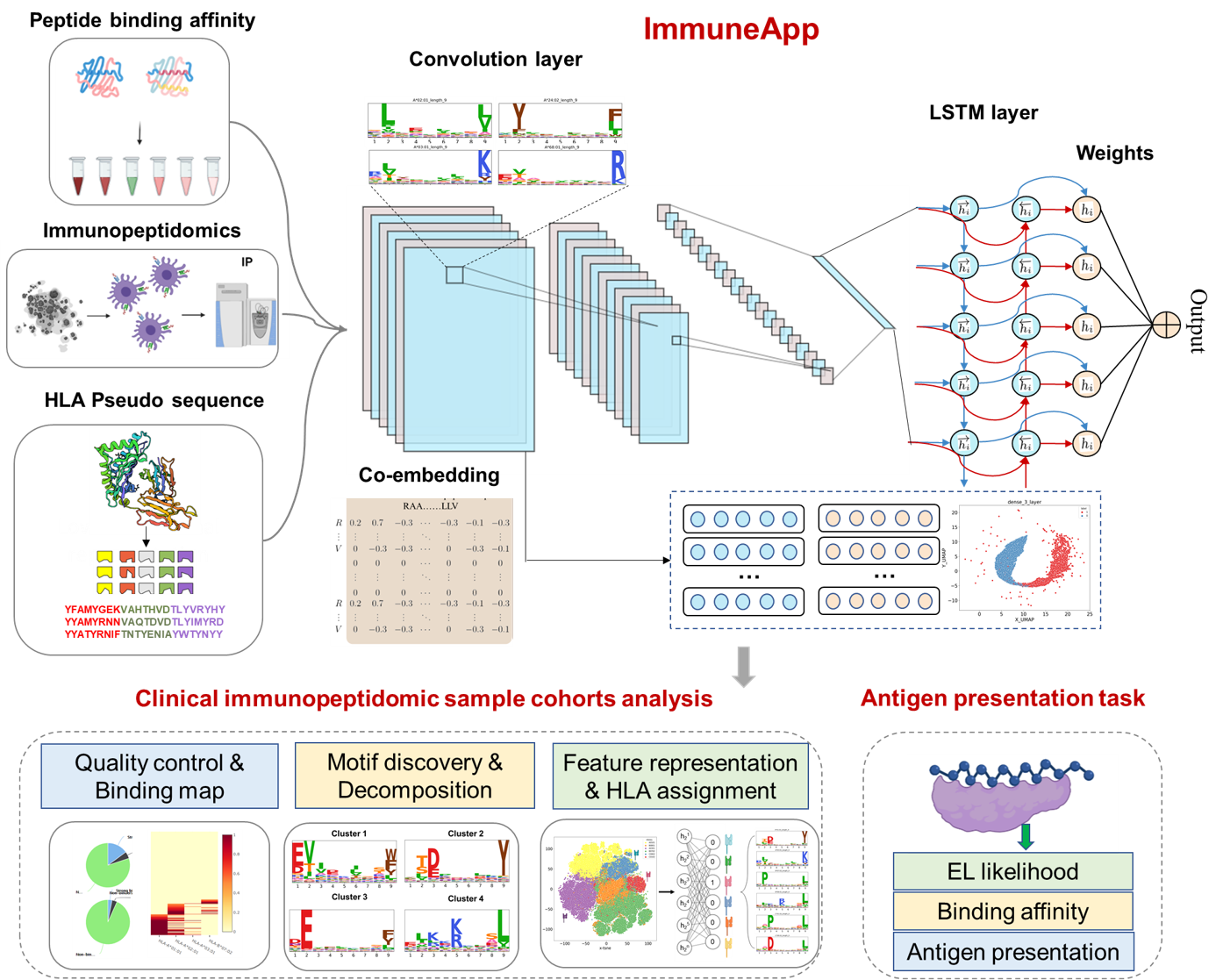
ImmuneApp application & performance
Our method was compared with NetMHCpan4.1 and MixMHCpred 2.2, which utilize NNalign-MA and MixMHCp for the deconvolution of immunopeptidomics data, respectively. We curated a dataset of 435,397 eluted ligands covering 86 HLA alleles from 47 recently published samples. Initially, AUROC, AUPRC and PPV were computed to evaluate the capacity of predictors in recognizing true ligands within extensive random peptide libraries from the human proteome. The results, illustrated in Figure 3B and Figure S5, indicated that our approach improves EL predictive capability, reaching mean AUROC of 0.9650 and mean AUPRC of 0.7600 when stratifying by samples. By comparison, NetMHCpan-4.1 produced a mean AUROC of 0.9155 and a mean AUPRC of 0.6071. Similarly, MixMHCpred 2.2 attained a mean AUROC of 0.9029 and a mean AUPRC of 0.6328. Significantly, the PPV values across samples calculated for each method are 0.8747 (our approach), 0.7689 (NetMHCpan-4.1) and 0.7970 (MixMHCpred-2.2), respectively, demonstrating our method is more sensitive to retrieve HLA-bound peptides observed in patient-derived tumor cell lines. In addition, we also expanded our evaluation by applying a more granular stratification (stratify by both sample and peptide length). Our method again outperformed other two tools, which yields mean AUROC, AUPRC and PPV of 0.9239, 0.6410, and 0.7913, respectively. By comparison, NetMHCpan-4.1 produced a mean AUROC of 0.8550, a mean AUPRC of 0.5080 and a mean PPV of 0.6750. Similarly, MixMHCpred 2.2 attained a mean AUROC of 0.8518, a mean AUPRC of 0.5367 and a mean PPV of 0.7103. Compared to two well-established and widely used tools trained on immunopeptidomics data, our approach constitutes the 8.06%, 19.43% and 11.40% improvement in AUROC, AUPRC and PPV, respectively.
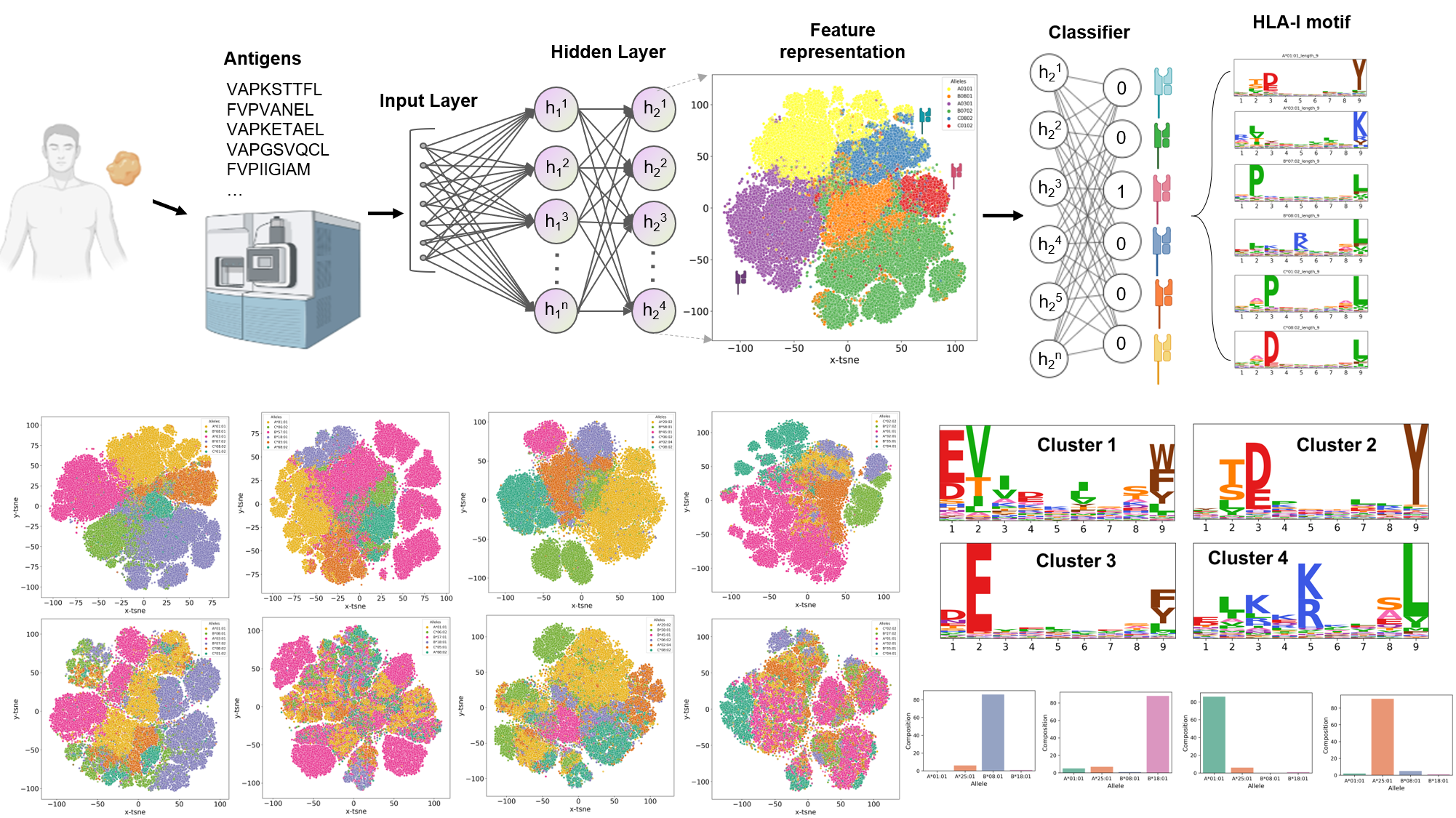
Application to patient cancer immunopeptidome
We analyze multiple melanoma-associated immunopeptidomics datasets. ImmuneApp accurately provide motif reconstruction and decomposition and other tasks.
More>>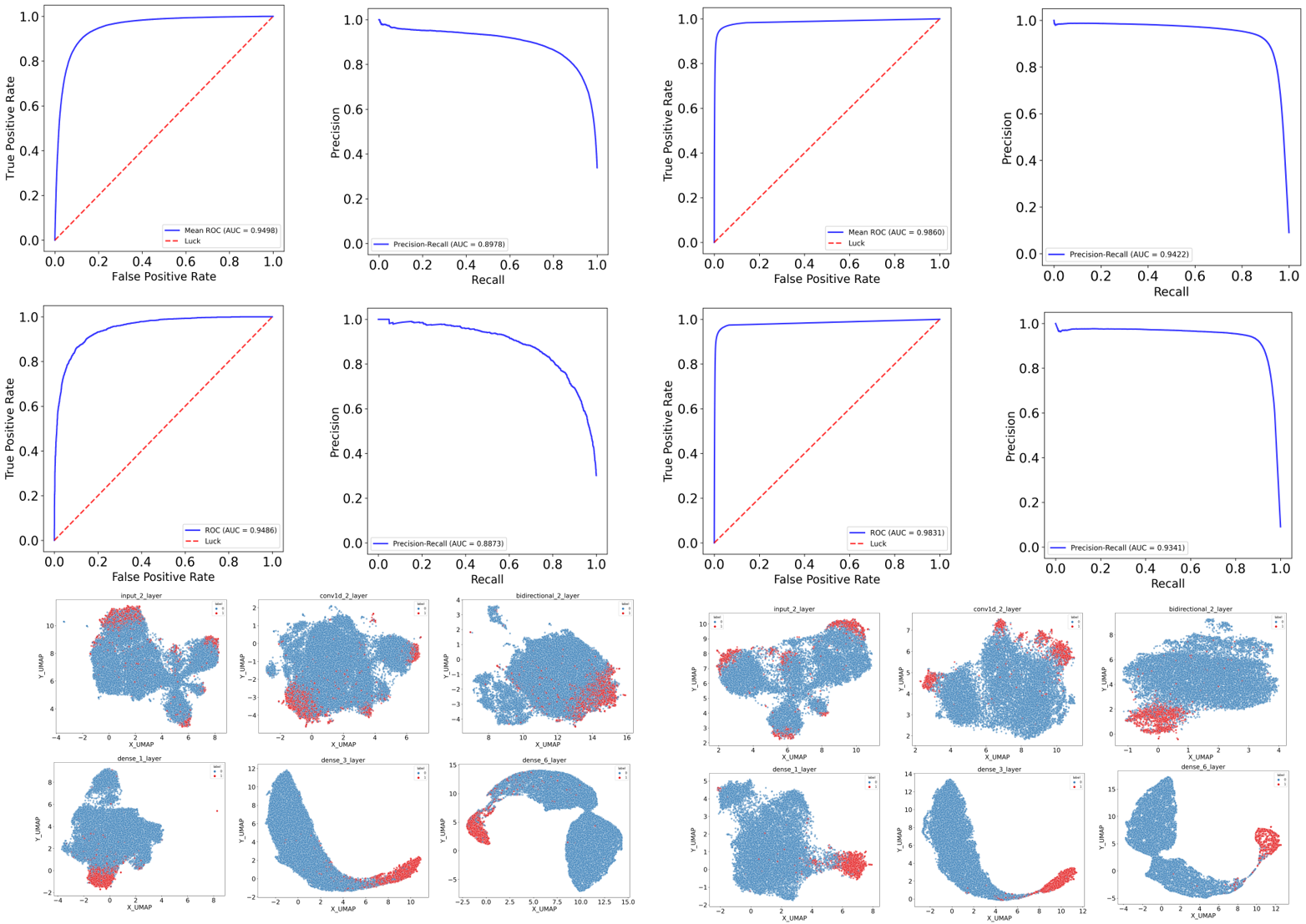
Cross-validation and model visualization
ImmuneApp had high performance in both training set and independent dataset. In addition, the learned features turn into more and more discriminative along the layer hierarchy of the model.
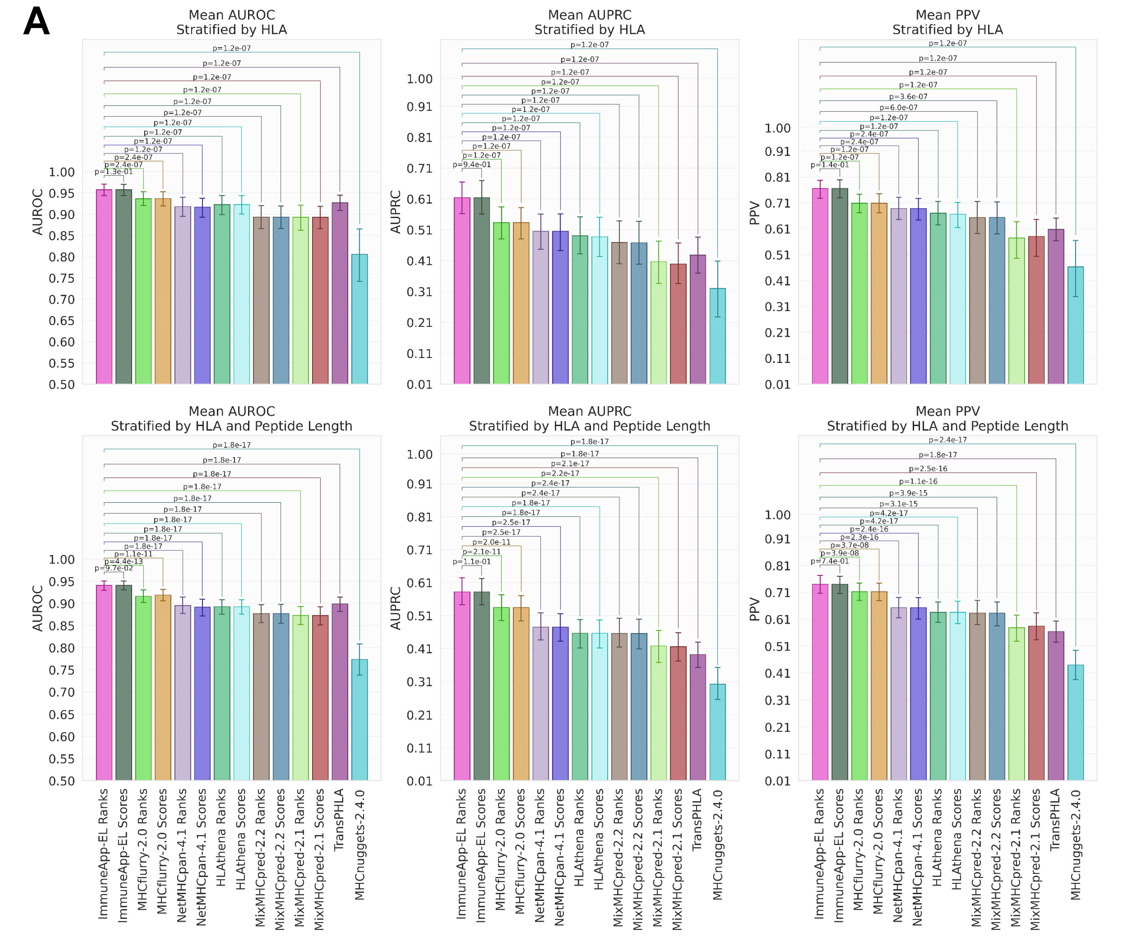
Performance comparison
Our improved deep learning framework outperforms existing methods in validation datasets in terms of both overall accuracy and PPV.
Developers:
Haodong Xu, Ruifeng Hu, Zhongming Zhao @ CPH, UTHealth-Houston SBMI.
ImmuneApp is free and open to all users and there is no login requirement.