1. Introduction
The presentation of peptides bound to major histocompatibility complex (MHC) proteins on the surface of antigen-presenting cells and subsequent recognition by T-cell receptors is fundamental to the mammalian adaptive immune system. Therefore, accurate predictions of the peptides presented on HLA alleles is important to study T cell immunity. Mass spectrometry (MS)-based immunopeptidomics is maturing into an automatized, high-throughput technology, producing large-scale datasets of clinically relevant MHC class I-associated peptides. While it provides us with more data to train higher-performance models, the development of quality control and downstream analysis tool is important for instrument operators and scientists in charge of downstream data interpretation. In this study, we developed the ImmuneApp, a multimodal CNN-LSTM Neural Network for predicting the likelihood of antigen presentation in the context of specific HLA class I alleles, which could provide binding measurements, eluted ligand likelihood and overall antigen presentation prediction. We further expanded ImmuneApp into a semi-automated online tool for personalized analysis of large clinical immunopeptidomic sample cohorts and numerical scores for QC analysis of tumor biopsies, which could perform quality control, motif discovery (unsupervised and supervised) and decomposition, HLA assignment and antigen presentation prediction for clinical immunopeptidomic sample cohorts.
2. Model
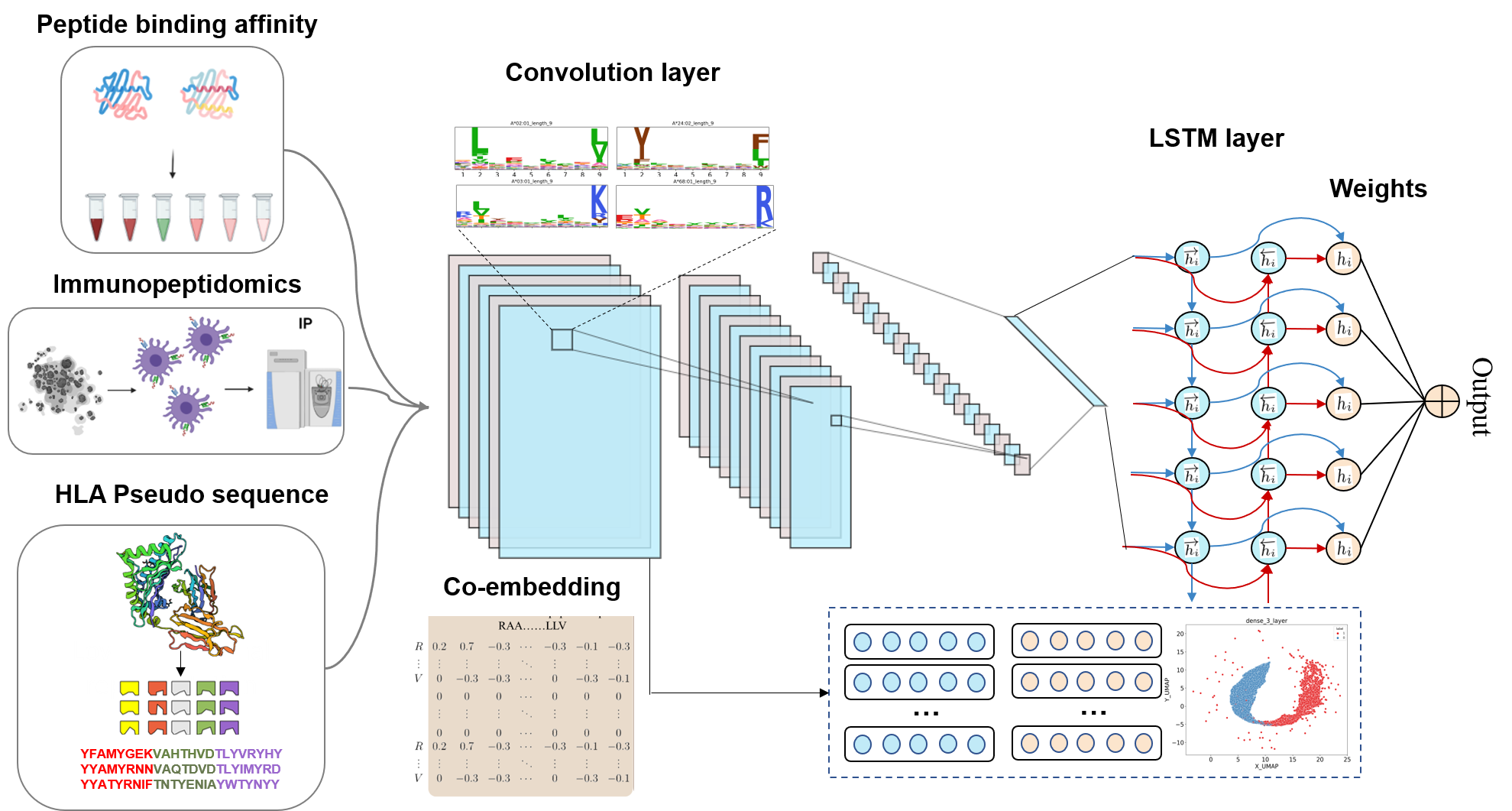
Figure 1. General pipeline for building models.
2.1. Model training
The ImmuneApp 1.0 predictor is a new pan-allele MHC class I binding predictor that supports variable-length peptides up to 13-mers. The ImmuneApp model builds upon the convolutional neural network-long short term memory (CNN-LSTM) hybrid architecture by incorporating more than 900,000 quantitative Binding Affinity (BA) and Mass-Spectrometry Eluted Ligands (EL) peptides by automatically learning discriminative features and essential residues from the ligand sequences along the layer hierarchy. The input to each neural network consists of (1) an encoding of the peptide amino acid sequence and (2) an encoding of the amino acids at 34 selected positions from a multiple sequence alignment of a large number of MHC class I alleles on the BLOSUM50 substitution matrix. A rectifier activation convolution layer transforms the input matrix into an output matrix with a row for each convolution kernel and a column for each position in the input (minus the width of the kernel). Each kernel is effectively a sequence motif. Max pooling downsamples the output matrix along the spatial axis, preserving the number of channels was used for encoding MHC class I alleles. The subsequent recurrent layer contains LSTM units connected end-to-end in both directions to capture spatial dependencies between motifs. Recurrent outputs are densely connected to a layer of rectified linear units. The activations are likewise densely connected to a sigmoid layer that nonlinear transformation to yield a vector of probability predictions of the likelihood of antigen presentation in the context of specific HLA class I alleles.
2.2. Model performance
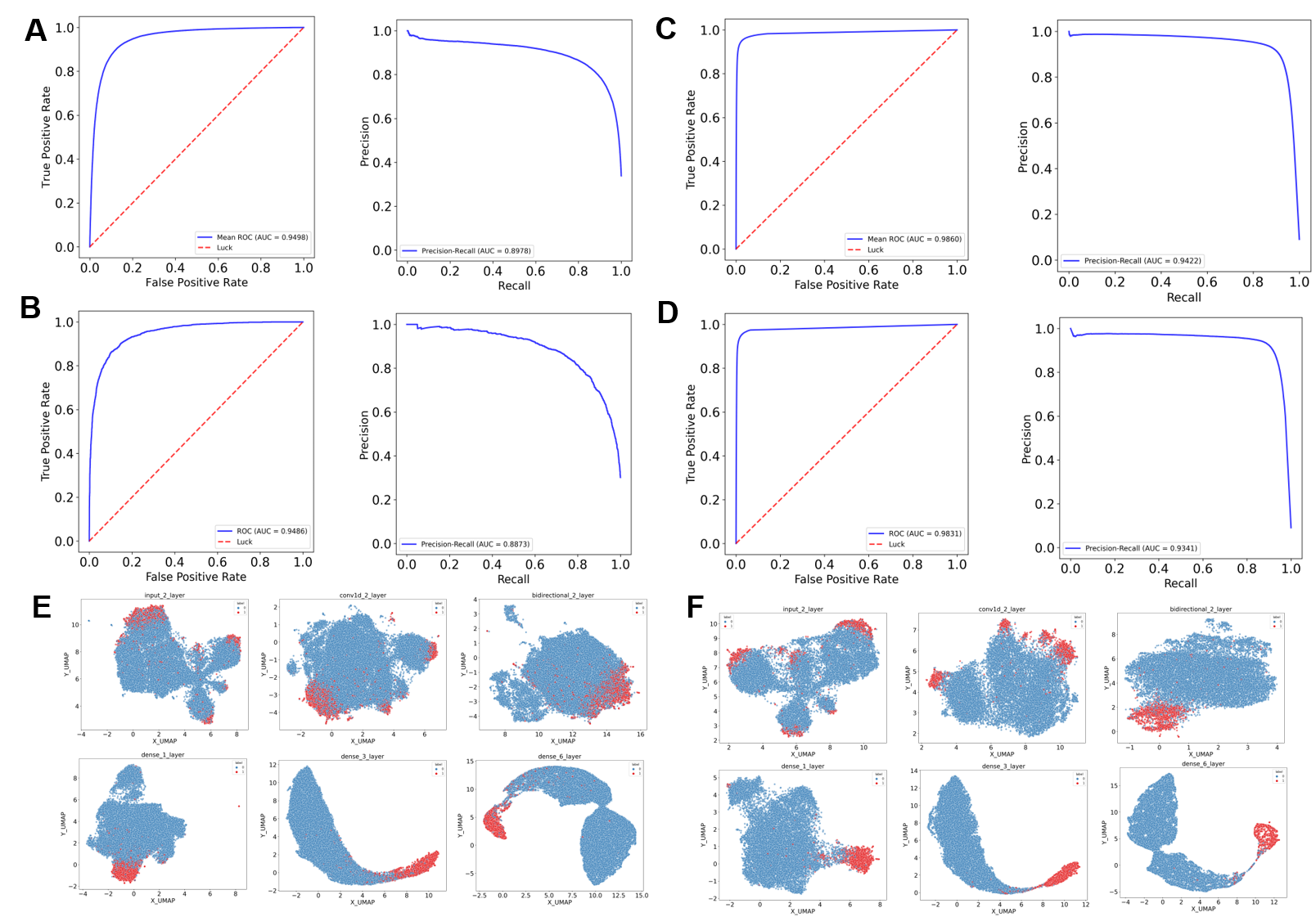
Figure 2. Five-fold cross-validation and the excellent interpretability of the model.
To evaluated the accuracy and robustness of ImmuneApp, we performed cross validation on the training data sets. We found that ImmuneApp had high performance in both training set and independent dataset of quantitative binding affinity and mass-spectrometry eluted ligands. Moreover, we visualized the ligands and random peptides using UMAP method based on the feature representations uncovered at different network layers. The learned features turn into more and more discriminative along the layer hierarchy, with ligands and random peptides being mixed at the input layer without clear boundary, culminating with a clear separation in the output layer..
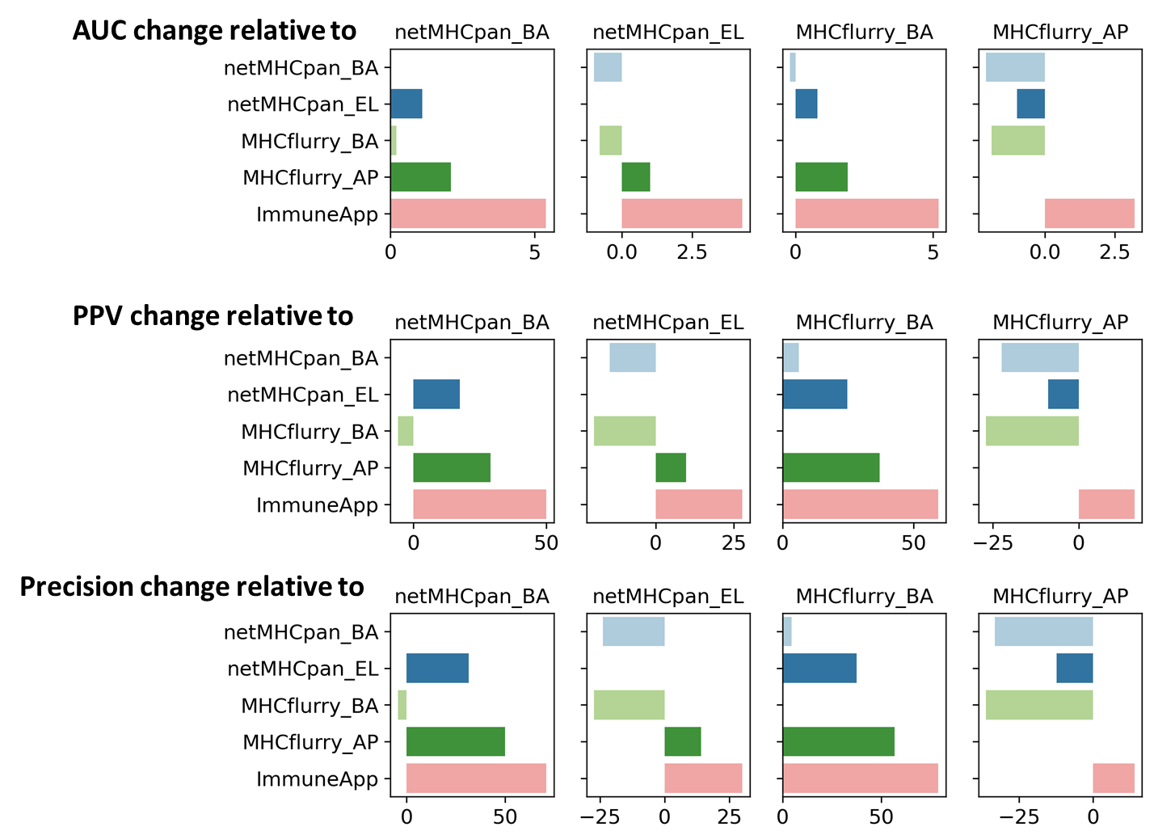
Figure 3. Tools comparision by three different metrics.
To further evaluate predictive power at distinguishing MS hits from decoys, we constructed datasets consisting of the observed allele- and length-specific binders in our MS data (n) along with 99 × n random decoys from the human proteome and considered the fraction of correctly predicted binders in the top 1% of the dataset and then calculate the positive predictive value (PPV) for comparison. The PPV, as defined here, focuses on a predictor’s ability to rank hits far above the decoys. To calculate PPV, for each sample we sorted the n hits and 99n decoys by their predictions, and calculated the fraction of the top n peptides that are hits. A random predictor would score 0.01 in PPV. We compared the PPV scores of ImmuneApp with two widely used tools (netmhcpan4.0 and mhcflurry 2). The results show that our model outperforms existing methods, with improvements of PPV scores range from 15%-60%..
2.3. Model application
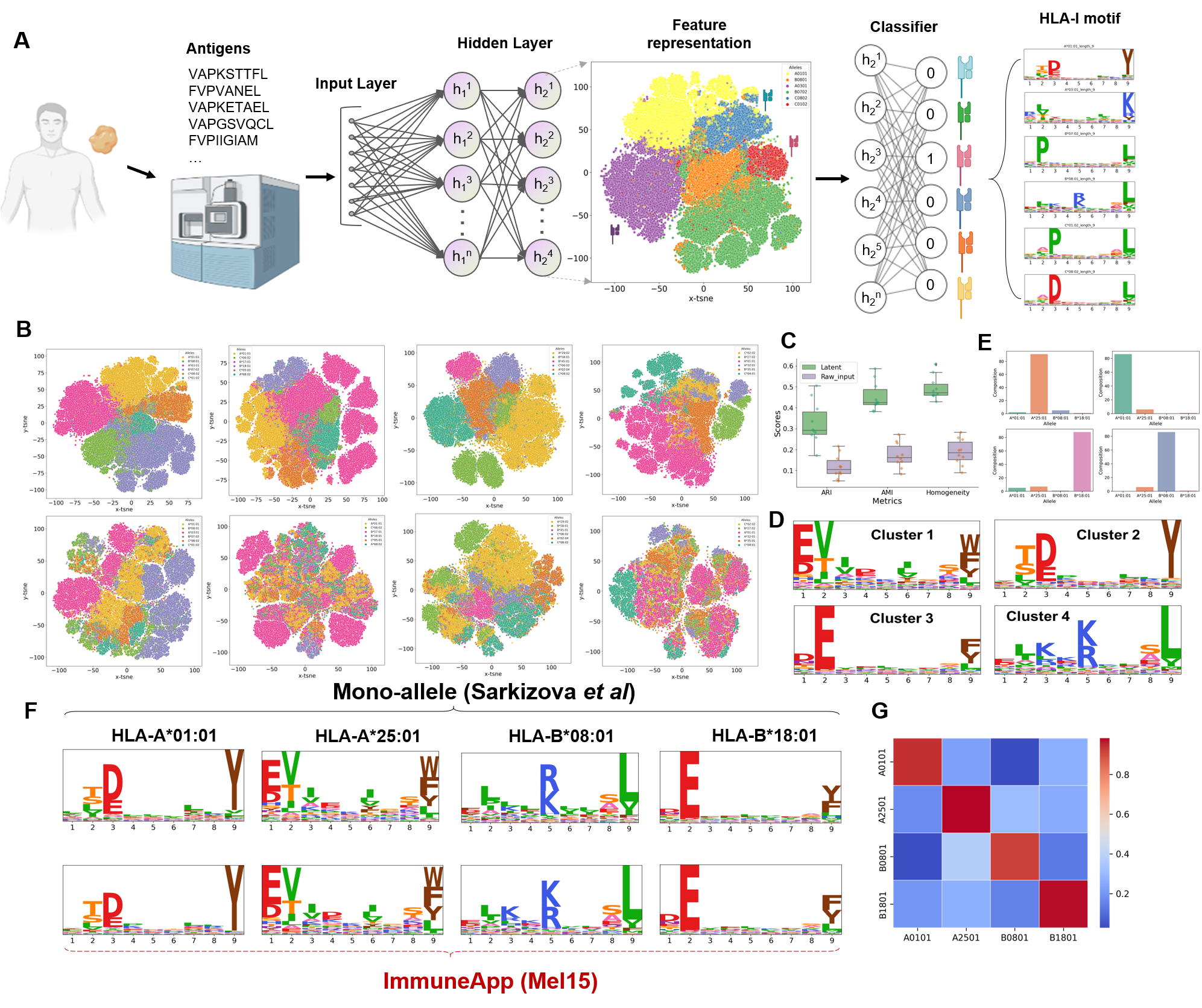
Figure 4. Model application on clinical immunopeptidomic sample.
To assess the relevance of our approach in a clinical setting, we analyze melanoma-associated immunopeptidomics datasets. We found the deep learning model provides a lower-dimensional representation of peptides with a clear interpretation in terms of associated HLA type. And ImmuneApp accurately provide motif reconstruction and decomposition and other tasks.
3. Webserver workflow
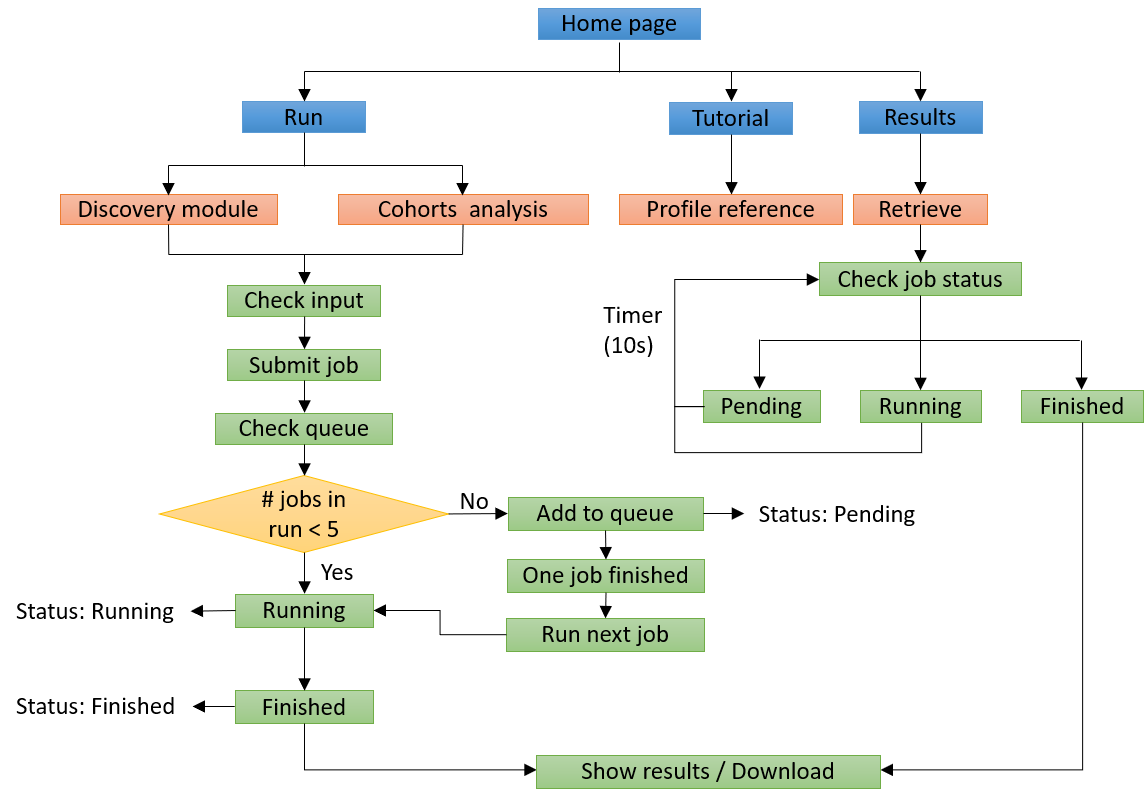
Figure 4. Workflow of the webserver.
Our webserver provides user-friendly interfaces for users to submit jobs, check job status, and retrieve results.
4. Prediction Portal
Prediction Portal provides antigen presentation prediction and epitope discovery by multimodal deep neural network.
4.1. Input
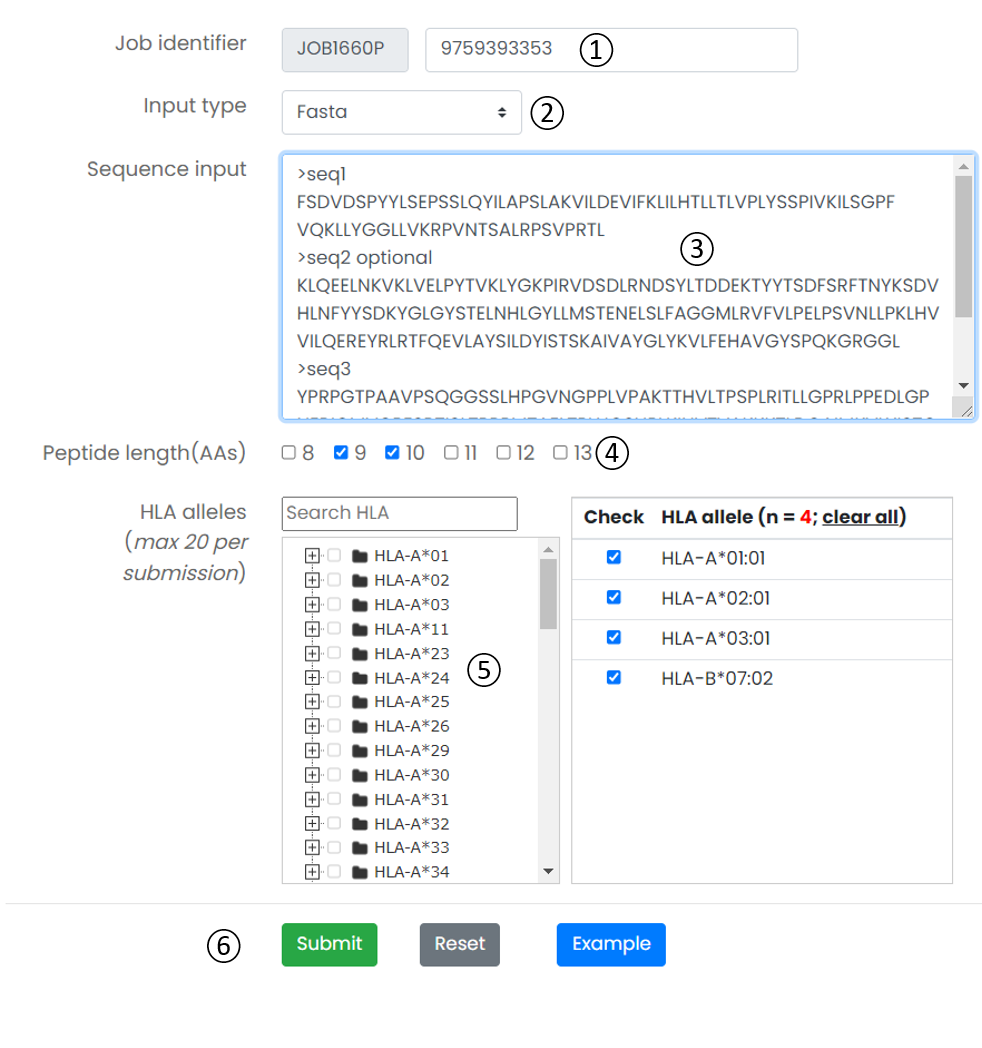
Figure 5. Job submission form.
- Job identifier: Job identifier can be generated automatically or customized by the submitter. It is confidential to other users and can be used for job status monitoring and result retrieval.(See Results page).It is required.
- Input type: Provides two input formats, including the classic protein FASTA format and direct input of multiple peptides.
- Input textarea: The user can directly copy the protein sequence or peptide data in the input box.
- Peptide length(AAs): When the input method is Fasta format. The user needs to select one or more peptide lengths so that the server can construct a library of candidate antigen peptides.
- HLA alleles: The ImmuneApp 1.0 server predicts peptides binding to more than 10,000 human MHC molecule. We constructed a classification tree of HLA. Users can quickly retrieve and submit candidate HLA alleles through the search box and tree map. Each submitted task is allowed to select up to 20 HLA alleles.
- Operation buttons: Submit, reset the submission form, or access the example dataset.
4.2. Output
Predicted antigen presentation
In addition to in vitro binding measurements prediction task, The ImmuneApp 1.0 server also provides eluted ligand likelihood prediction and overall antigen presentation prediction. The ImmuneApp 1.0 server will first return a statistical table based on the results of eluted ligand likelihood prediction. It includes the number of candidate antigen peptide libraries constructed in each sample (protein), the number and proportion of peptides presented by different HLA alleles. ImmuneApp also provides detailed predicion results, which includes the score, ranking and binding strength of all candidate antigen peptides presented by different HLA alleles in each sample (protein).
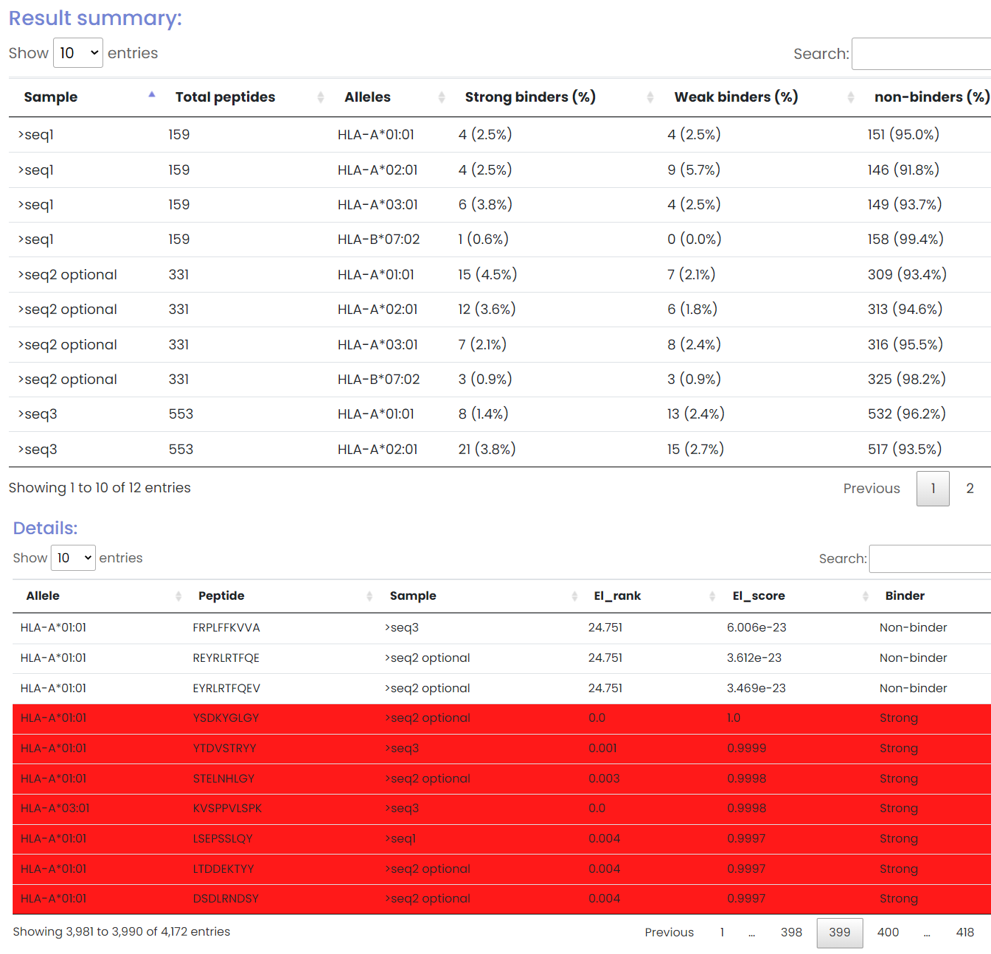
At the page bottom, a “download” button is provided for downloading all the results.
5. Analysis portal (immunopeptidomic cohorts analysis)
The analysis portal is a semi-automated online tool that enables rapid assessment and analysis of single or multiple MHC class I immunopeptidomic datasets, including datasets generated from large clinical sample cohorts. Users can upload multiple immunopeptidomic datasets, and then select alleles annotated by the HLA typing tool. ImmuneApp 1.0 will return multi-level analysis results, including:
- Quality control
- Binding map visualization
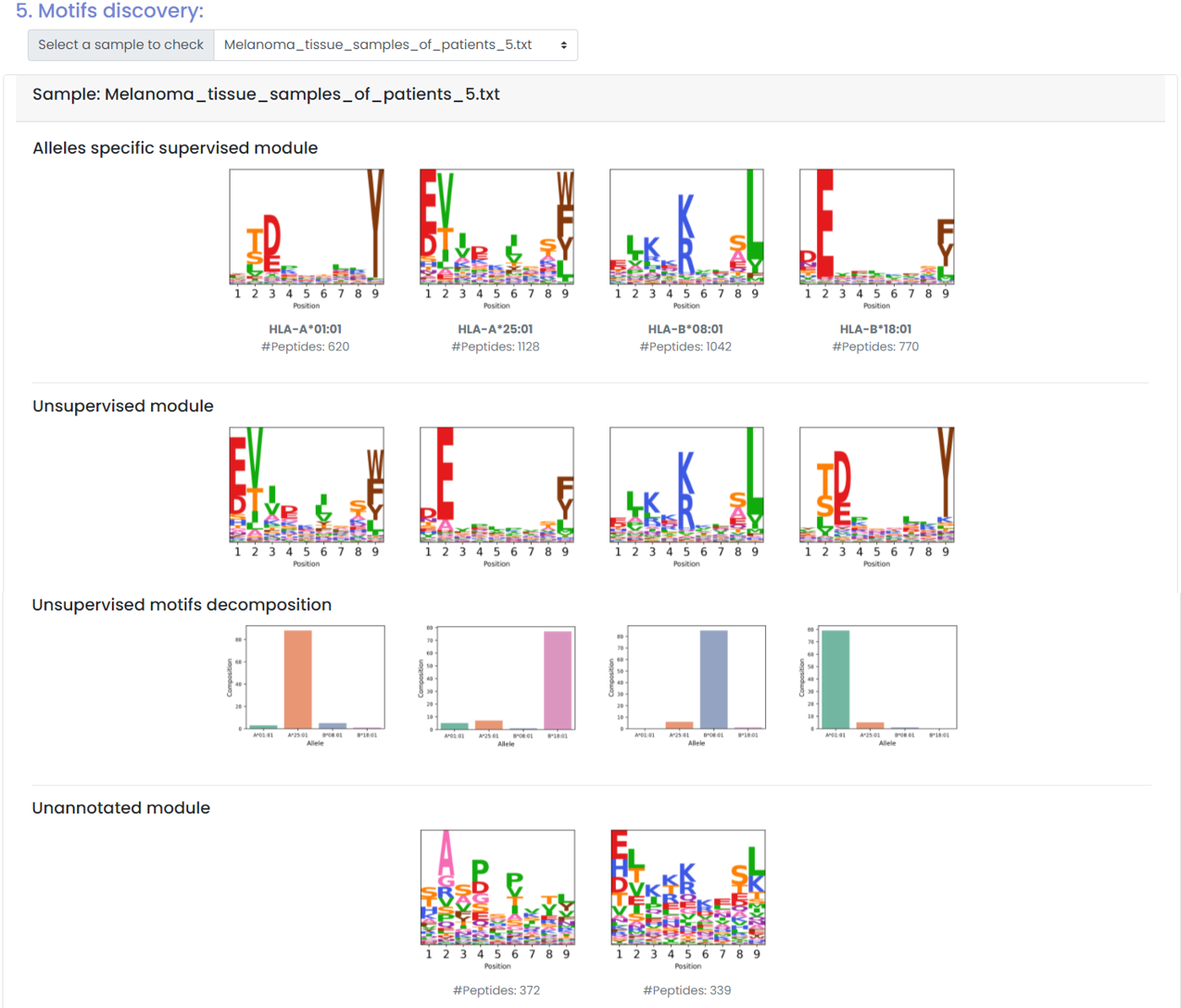
- Motif discovery (unsupervised and supervised)
- Motif decomposition
- HLA assignment
- Antigen presentation prediction
5.1. Input
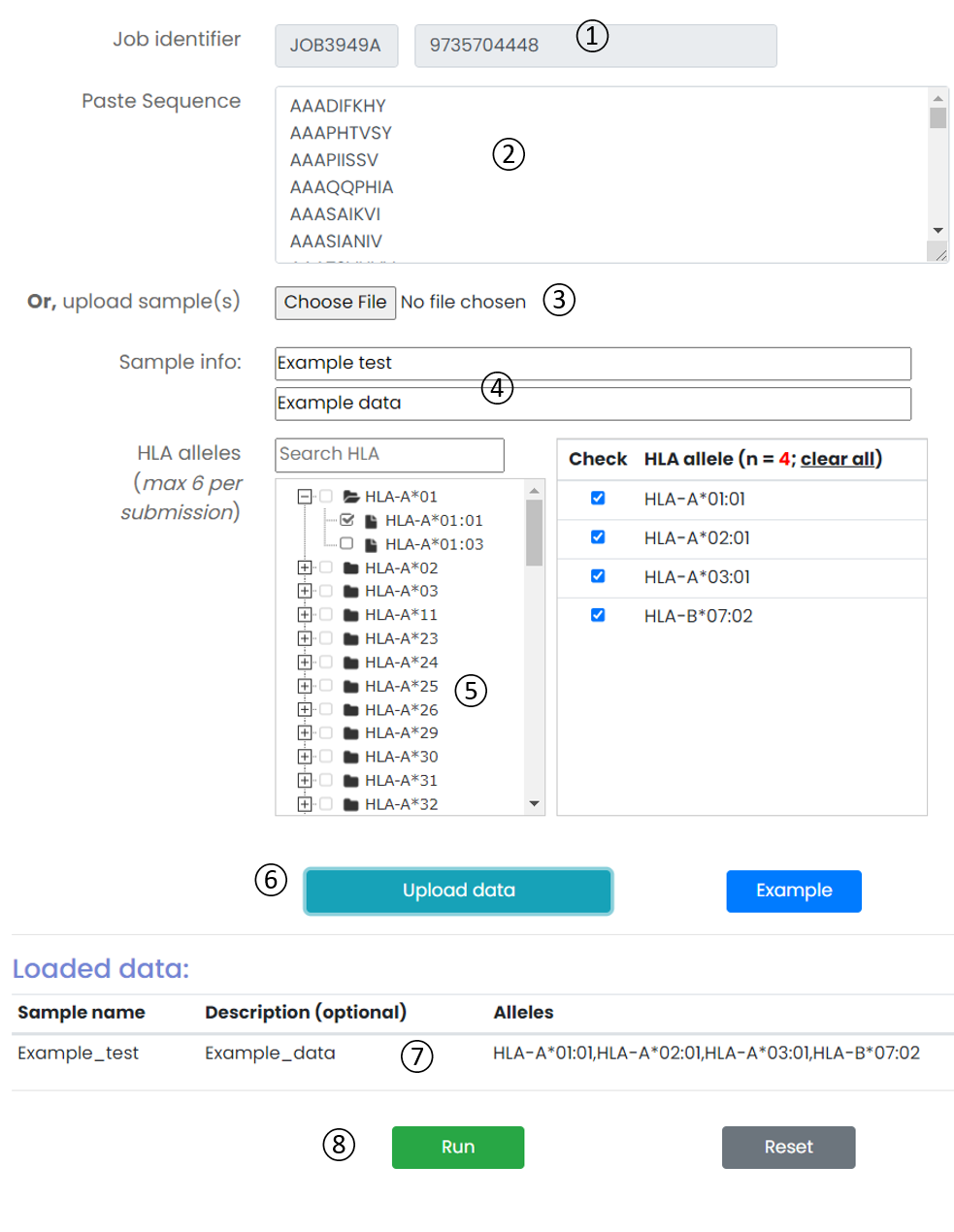
Figure 5. Job submission form.
- Job identifier: Job identifier can be generated automatically or customized by the submitter. It is confidential to other users and can be used for job status monitoring and result retrieval.(See Results page).It is required.
- Input textarea: The user can directly copy immunopeptidomic cohorts sample data in the input box.
- Upload sample(s): The user can also upload immunopeptidomic cohorts sample to the server.
- Sample info: The user needs to provide identification information for each sample.
- HLA alleles: The ImmuneApp 1.0 server predicts peptides binding to more than 10,000 human MHC molecule. We constructed a classification tree of HLA. Users can quickly retrieve and submit candidate HLA alleles through the search box and tree map. Each submitted task is allowed to select up to 6 HLA alleles.
- Operation buttons: Upload immunopeptidomic cohorts sample to the server by this button
- loaded data: A list of immunopeptidomic cohorts uploaded by users for analysis.
- Operation buttons: Submit, reset the submission form, or access the example dataset.
5.2. Output
(1) Quality control and binding annotations

(2) Antigen presentation prediction and globle binding map (HLA assignment)
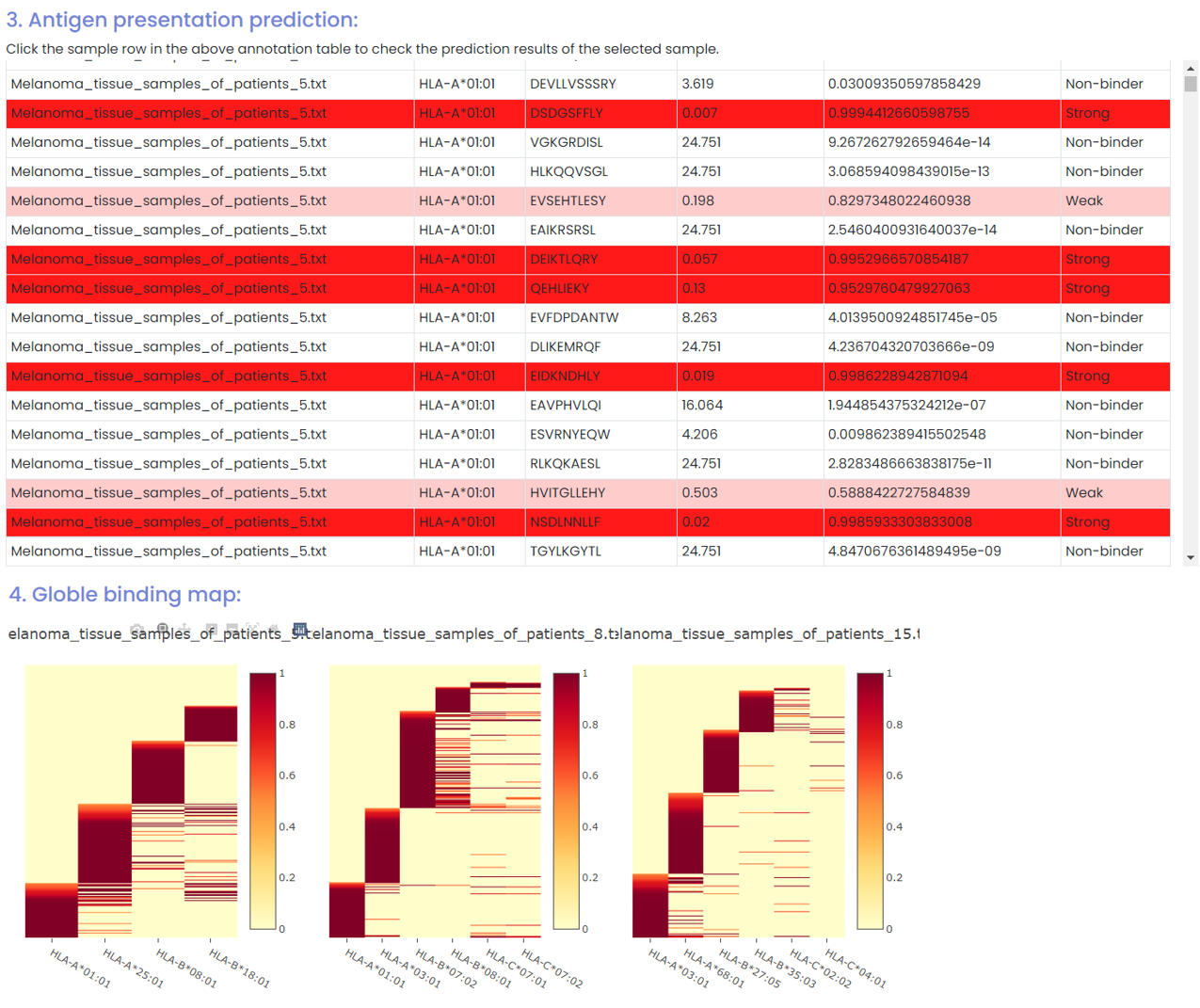
(3) Motif discovery and decomposition
At the page bottom, a “download” button is provided for downloading all the results.